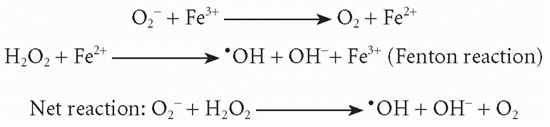This chapter focuses on infections by intracellular bacteria with emphasis on both pathogenicity and general immune mechanisms underlying protection. Intracellular bacteria comprise numerous pathogens, some of which are of utmost medical importance. Ancient (but still existent), as well as newly emerging, diseases are caused by intracellular bacteria, with tuberculosis (TB), typhoid, leprosy, and trachoma as the most relevant etiologically associated infections.
Intracellular bacteria live inside host cells for most of their lives. This coexistence must allow survival of the infected cell; therefore, intracellular bacteria generally lack toxicity. Instead, they have evolved multiple strategies to interfere with key host cell biologic processes to promote replication or persistence. This has direct consequences for the immune response evoked by the host attempting to resolve intracellular infection. In fact, acquired resistance against intracellular bacterial infections depends on activation of multiple arms of the immune system. In this chapter, we start by examining the complex molecular crosstalk between pathogen and host. We then consider how these molecular events translate into the development of adaptive immunity. Finally, we examine effects of both adaptive and innate immune mechanisms on long-term effects of intracellular bacterial infection. Such long-term effects include tissue remodelling, nonresolved inflammation, and chronic immune responses, which for the most part are tolerated by the host. We hope this chapter will stimulate an interest in intracellular infection and the unique immunologic insights it can provide.
FEATURES OF INFECTIONS WITH INTRACELLULAR BACTERIA
Intracellular Bacteria/Public Health Relevance
Bacterial pathogens are prokaryotic microorganisms that cause disease in a given host species. They are single cells, typically micrometers in length, and present a specific spectrum of interactions with their human host. Infectious disease is the direct and invariable consequence of an encounter between host and pathogen. Often, it is the eventual outcome of complex interactions between them. There is a marked degree of diversity among bacterial species that are able to induce intracellular infections. This encompasses structural organization of the cell wall (ie, gram-negative versus -positive, salmonellae versus listeriae), presence of motility organelles (eg, fimbriae and flagella, of salmonellae and listeriae1,2) or secretion apparatus (type III secretion system [T3SS], type IV secretion system (T4SS), type VII secretion system [T7SS] in Salmonella,3Legionella,4 and Mycobacteria,5 respectively), and metabolic characteristics that dictate replication time (fast- versus slow-growing, Salmonella versus Mycobacterium). This diversity is reflected most often in differences related to cell biology of the pathogen. However, major patterns of the immune response are shared, as detailed in the following.
Of paramount significance for humans are Mycobacterium tuberculosis, Mycobacterium leprae, Salmonella enterica serovar Typhi, Chlamydia trachomatis, and the etiologic agents of TB, leprosy, typhoid, and trachoma, respectively, which together afflict more than 200 million people globally. Some opportunistic pathogens such as Mycobacterium avium/Mycobacterium intracellulare are gaining increasing significance particularly for immunodeficient patients, such as acquired immunodeficiency syndrome (AIDS) sufferers. Many zoonotic agents are intracellular bacteria and include Chlamydia psittaci (psittacosis), Brucella (brucellosis, Malta fever/Bang disease), Coxiella burnetii (Q fever), Rickettsia (Rocky Mountain/Mediterranean spotted fever), Francisella tularensis (tularemia), Burkholderia mallei (glanders), Yersinia pseudotuberculosis (yersiniosis), Yersinia pestis (plague), Bartonella (cat scratch disease), and Listeria monocytogenes (listeriosis). Other opportunistic pathogens with an intracellular lifestyle that are relevant for human health include Legionella pneumophila (Legionnaires disease) and Ehrlichia (ehrlichiosis).
Epidemiology and Pathogenesis
Some intracellular bacteria, in particular Rickettsia, are introduced directly into the bloodstream by insect bites from where they have ready access to internal tissues. Most intracellular bacteria, however, enter the host through the mucosa.6 Major ports of entry are the lung for airborne pathogens, such as M. tuberculosis and L. pneumophila, and the intestine for foodborne pathogens, such as S. enterica and L. monocytogenes. Subsequently, intracellular bacteria pass through the epithelial layers. Either they actively induce transcytosis (ie, endo- and exocytosis) through the epithelial cells or they are passively translocated within phagocytes. Bacteria may be removed by nonspecific defense mechanisms such as mucociliary movements and gut peristalsis, or they may be destroyed by professional phagocytes without necessitating the specific attention of the immune system. Cells that survive these nonspecific defense reactions colonize deeper tissue sites and stably infect a suitable niche. At this stage, the host generally develops a specific acquired immune response.
BOX 40.1. CENTRAL FEATURES OF INFECTIONS WITH INTRACELLULAR BACTERIA
Infection is separated from disease, and the immune response is already induced at the stage of infection.
Infection persists latently in the face of dynamic interactions between pathogen and immune mechanisms.
The host-pathogen relationship represents a highly sophisticated form of parasitism that does not necessarily lead to disease but rather allows for long-lasting coexistence.
Infection includes the potential to harm the host severely at a later stage, and pathogenesis is strongly influenced by the immune response.
Infection is unsuccessful when the immune system succeeds in eliminating the pathogen before overt clinical disease develops. Alternatively, tissue damage increases to a significant level before the immune system succeeds in controlling the pathogen effectively and clinical disease develops. This is the case with many extracellular bacteria and is due to their cell lytic strategies (toxins) that cause diseases of acute type, but is less common for intracellular bacteria. Finally, it is possible that the immune response restrains the infectious agent but fails to completely eradicate it. Under these conditions, a long-lasting equilibrium between microbial persistence and the immune response unfolds. This balance, however, remains unstable and can be tipped in favor of the pathogen at a later time, converting infection into disease.
The time lapse between host entry and expression of clinical disease is termed incubation time and from what has been said previously, it follows that in many intracellular bacterial infections the incubation times are long-lasting to lifelong. By improving the immune response or by impairing bacterial growth (typically accomplished by chemotherapy), or both, disease can be overcome. Ideally, bacterial eradication is achieved; alternatively, some dormant bacteria continue to persist in niches poorly accessible to the immune response (Box 40.1).
There is a close correlation between the cell biology and cell tropism of the pathogen and disease signs and pathology in, for instance, enteric versus lung conditions. However, even bacteria that use the same niches as habitat show major clinical differences. This is due to pathogenintrinsic biology (ie, metabolism) and peculiarities of each species to subvert host elimination. For instance, M. tuberculosis, L. pneumophila, F. tularensis, and C. burnetii cause aerogenic infections with Coxiella being the most successful in establishing infection (one bacterium is sufficient to induce disease, probably the most infectious bacterium). These pathogens successfully infect lung macrophages and parenchymal cells to establish infection. However, TB has a protracted course and insidious clinical signs while francisellae and coxiellae induce acute and subacute pneumonias with tissue changes substantially different from TB. Disease pathogenesis is principally the outcome of the crosstalk between host and pathogen at the cellular level with strong influences from bacteria-specific behavior, while clinical signs and pathology are often induced by host responses.
“Idealized” Intracellular Bacterium
Although this chapter focuses on general mechanisms underlying immunity to intracellular bacteria, it is important to emphasize that this group is extremely heterogeneous. Therefore, the major hallmarks of intracellular bacterial infections will first be described for a nonexistent “idealized” intracellular bacterium (Table 40.1). Subsequently, characteristics of selected intracellular bacteria will be specified.
Facultative and Obligate Intracellular Bacteria
With respect to preferred habitat, intracellular bacteria can be divided into two groups: First, those pathogens that do not essentially depend on the intracellular habitat, including M. tuberculosis, Mycobacterium bovis, M. leprae, S. enterica, Brucella, L. pneumophila, L. monocytogenes, and F. tularensis(Table 40.2).7,8,9,10,11,12,13,14,15 Although these pathogens favor mononuclear phagocytes (MPs) as their habitat, other types of host cells are infected as well. M. leprae, for example, lives in numerous host cell types, notably in Schwann cells and hepatocytes serve as an important reservoir for L. monocytogenes. Although M. tuberculosis can infect a variety of mammalian cells in vitro, in vivo it seems to restrict itself to phagocytes and perhaps epithelial cells.
TABLE 40.1 Hallmarks of Intracellular Bacterial Infections
Hallmark 1
The intracellular lifestyle represents the distinguishing feature of intracellular bacteria.
Hallmark 2
T cells are the central mediators of protection against intracellular bacterial infections. In contrast, antibodies play a facilitating role.
Hallmark 3
Infections with intracellular bacteria are accompanied by delayed-type hypersensitivity, which expresses itself after local administration of soluble antigens as a delayed tissue reaction mediated by T cells and effected by macrophages.
Hallmark 4
Tissue reactions against intracellular bacteria are granulomatous. Protection against, as well as pathology caused by, intracellular bacteria are centered on these lesions. Rupture of a granuloma promotes bacterial dissemination.
Hallmark 5
Intracellular bacteria express little or no toxicity for host cells by themselves, and pathology is primarily a result of immune reactions, particularly those mediated by T-lymphocytes.
Hallmark 6
Intracellular bacteria coexist with their cellular habitat for long periods of time. A labile balance develops between persistent infection and protective immunity, resulting in long incubation time and in chronic disease. Accordingly, infection is dissociated from disease.
Hallmarks 1 to 4 should be considered essential, and Hallmarks 5 and 6 conditional, criteria for defining intracellular bacteria.
TABLE 40.2 Major Infections of Humans Caused by Facultative Intracellular Bacteria
Pathogen
Disease
Preferred Target Cell
Preferred Location in Host Cell
Preferred Port of Entry
Mycobacterium tuberculosis
Tuberculosis
Macrophages
Early phagosome
Lung
Mycobacterium leprae
Leprosy
Macrophages, Schwann cells, other cells
Phagolysosome, cytosol
Nasopharyngeal mucosa
Yersinia pestis
Plague
Macrophages
Autophagosome
Gut, skin
Salmonella enterica serovar Typhi
Typhoid fever
Macrophages
Late phagosome, spacious phagosome
Gut
Shigella spp.
Shigellosis
Macrophages
Cytosol
Gut
Brucella spp.
Brucellosis
Macrophages
Early phagosome
Mucosa
Legionella spp.
Legionnaires’ disease
Macrophages
Endoplasmic reticulum-derived phagosome
Lung
Listeria monocytogenes
Listeriosis
Macrophages, hepatocytes
Cytosol
Gut
Francisella tularensis
Tularemia
Macrophages
Late endosome, cytosol
Skin, lung, mucosa
The second group includes so-called obligate intracellular bacteria, which fail to survive outside host cells. Most of these bacteria prefer nonprofessional phagocytes as their habitat—for example, endothelial and epithelial cells. Rickettsiae, chlamydiae, and ehrlichiae are representatives of this group. They include Rickettsia prowazekii, Rickettsia rickettsii, Rickettsia typhi, and Orientia tsutsugamushi (Rickettsia tsutsugamushi until 1995), the etiologic agents of louse-borne typhus, Rocky Mountain spotted fever, typhus, and scrub typhus, respectively.16,17,18,19 Various biovars of Chlamydia trachomatis, which are responsible for trachoma,20 conjunctivitis, urogenital infections, and lymphogranuloma venerum,21 as well as C. psittaci and Chlamydia pneumoniae, causative agents of psittacosis or rare types of pneumonia,22 respectively, also belong to this group (Table 40.3). Certain obligate intracellular bacteria, such as Ehrlichia and Anaplasma phagocytophilum, parasitize blood cells.23,24C. burnetii, the causative agent of Q fever, resides in macrophages and lung parenchymal cells.25,26
TABLE 40.3 Major Infections of Humans Caused by Obligate Intracellular Bacteria
Preferential living in macrophages does not depend on specific invasion mechanisms but rather on highly sophisticated intracellular survival strategies. Yet, most facultative intracellular bacteria express unique invasion factors, if only to cross epithelial layers. Selection of nonprofessional phagocytes as habitat essentially depends on invasion molecules whereas survival inside these cells is generally less hazardous.
CELL BIOLOGY
For intracellular bacteria, entry into host cells represents the central requirement for survival in, as well as elimination by, the host. Host cell-directed uptake, called phagocytosis, is a feature of the so-called professional phagocytes that comprise polymorphonuclear granulocytes (PNGs) and MPs. Examples of bacteria that are engulfed by phagocytosis include M. tuberculosis, L. pneumophila, and C. burnetii. Entry induced by the pathogen is termed invasion, which allows entry into nonphagocytic cells (nonprofessional phagocytes). Salmonellae, shigellae, and listeriae are paradigms of enteroinvasive pathogens.27 Contact between host cells and pathogens proceeds either directly via receptor-ligand interactions or indirectly via deposition on the surface of the pathogen of host molecules for which physiologic receptors exist on the target cell.
Depending on the cellular target, the final outcome of host cell entry varies markedly.
Nonprofessional phagocytes are nonphagocytic, and hence entry depends on expression of surface receptors that can be exploited for invasion. Because of their low antibacterial activities, they primarily serve as a habitat.
PNGs are short-lived. Because they are highly phagocytic and express potent antibacterial activities constitutively, uptake by PNGs is often fatal for the pathogen.
MPs are phagocytic and express medium to high antibacterial activities depending on their activation status. Accordingly, they serve both as habitat and as effector cell.
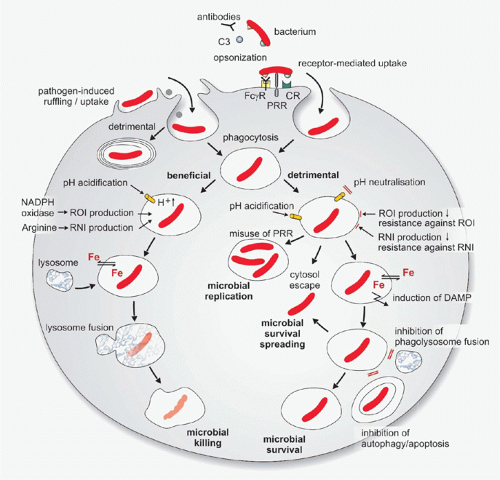
Following entry, bacterial pathogens begin intravacuolar life.28 Two main strategies are followed to avoid killing: 1) avoidance of delivery to degradative lysosomes, either by blocking phagosome maturation, divergence from the endocytic pathway to establish a vacuole with unique features, or by escape into the cytosol; and 2) development of strategies to survive within acidic degradative organelles. Certain bacteria have developed mechanisms that allow them to impede nutrient flow inside the infected cell for their own benefit, to modulate generation of antimicrobial molecules, or to alter cell death pathways.29,30 In the following, the major steps from uptake to bacterial elimination by, or survival in, host cells will be described (Fig. 40.1).
Adhesion and Invasion
Adhesion to mammalian cells is a prerequisite for extracellular colonization and for host cell invasion. Bacterial adhesins that solely expedite adhesion to host cells are expressed by numerous extracellular bacteria. In contrast, invasion-inducing molecules are a feature of bacteria that permanently or transiently enter host cells. Adherence to the cell membrane is based on protein-protein interactions mediated by adhesins, such as internalins (bind E-cadherin) of L. monocytogens or invasins (bind β1 integrins) from Yersinia. Adhesins may be located on the bacterial surface or on pili. In addition, adhesion may be induced by bacterial virulence factors, which recruit fibronectin to attach to host cells by binding to integrins.29,31
Although induced by the bacterium, invasion is ultimately a function of the host cell. Following adhesion, invasion can be induced in two ways. First, cell signaling by host cell receptors that serve as targets of adhesion induces uptake; second, uptake is induced independently from the molecules that mediate adhesion.31,32 The term “zipper mechanism” refers to the first process. Bacterial proteins interact with host cell surface proteins to mediate internalization. This term has been suggested for the highly selective receptor-mediated bacterial entry, whereas the term “trigger mechanism” has been proposed for indiscriminate, apparently adhesion-independent uptake. Bacterial effectors are delivered to the host cytosol via a secretion system to induce bacterial entry.27
Entry by Zipper Mechanisms
Host cell invasion by Yersinia and L. monocytogenes are examples of invasion via the “zipper mechanism.” Receptor binding induces phagocytic mechanisms in nonprofessional phagocytes similar to those that are constitutively operative in MPs. The eukaryotic cell membrane tightly enwraps the bacterium and a cascade of events, including protein phosphorylation, ubiquitination, and phospholipid modifications then contribute to vacuole genesis.28 Host entry of L. monocytogenes through the intestinal epithelia is mediated by internalin on the surface of this pathogen and E-cadherin on human epithelial cells.33 Murine E-cadherin does not serve as a receptor for internalin due to an amino acid substitution in position 16.34 Schwann cells, a major target of M. leprae, are shielded by a basal lamina composed of laminin, collagen, and proteoglycans. The unique tropism of M. leprae for peripheral nerves appears to be due to bacterial binding to laminin. This molecule, which serves as natural ligand for integrins, thus provides a link between pathogen and Schwann cell.35 Caveolin and lipid rafts serve as entry portals for Brucella and certain strains of Chlamydia. More recently, evidence was provided that clathrin-mediated endocytosis contributes to entry of L. monocytogenes and Rickettsia in a similar zipper mechanism.36,37 Septins, which are small guanine triphosphatases (GTPases), are able to form filaments and interact with actin to facilitate bacterial entry as well.38
Entry by Trigger Mechanisms
Different molecules and mechanisms participate in host cell entry by S. enterica. Interactions between S. enterica and host cells causes large “membrane ruffling” at the site of attachment followed by bacterial entry. Ruffling induces indiscriminate uptake even of other particles in the vicinity of S. enterica. This process has been termed macropinocytosis. S. enterica triggers its uptake by exploiting the signaling machinery of the host cell, thus inducing cytoskeletal rearrangements. In certain mouse cells, S. enterica induces phosphorylation of the receptor for the epidermal growth factor.39 Yet, S. enterica can also enter cells that do not express the epidermal growth factor receptor. This pathogen possesses two T3SS that allow it to directly manipulate intracellular molecules within host cells.40Salmonella outer proteins are secreted into the host cells rapidly after contact. Salmonella outer proteins activate the small GTP-binding protein cell division control (CDC)42 of the Ras superfamily, which, in turn, induces the reorganization of the actin cytoskeleton, thus promoting bacterial invasion through membrane ruffling. A homolog of Salmonella outer protein (termed Salmonella outer protein 2) performs similar functions, and hence the two molecules may partly compensate each other’s functions. The transiently intracellular pathogen Shigella utilizes similar mechanisms for uptake via membrane ruffling.41,42 More recently, filopodia were shown to trap and direct shigellae to target cells in a process that involves bacterial T3SS.43 Generally, modulators of the eukaryotic cell cytoskeleton are T3SS or T4SS products, which modulate GTPase cycling of proteins of the Rho, Rab, and Arf families.44
FIG. 40.1. The Multiple Encounters Between Phagocytes and Intracellular Bacteria. Bacterial uptake is both pathogen-induced and receptormediated. Multiple opsonic receptors, including complement receptor (CR) and receptors for immunoglobulins (FcγR), facilitate phagocytosis. In addition, certain pattern recognition receptors (PRRs) may contribute to bacterial internalization. Macrophages and neutrophils direct their bacterial cargo to the endosomal pathway, endosomes become acidic and progressively accumulate reactive oxygen intermediates (ROIs) and reactive nitrogen intermediates (RNIs). The phagosome maturation process culminates in fusion between late endosomes and lysosomes, which leads to bacterial killing but intracellular bacteria can interfere with phagosomal killing. Certain bacteria modulate early uptake events to prevent endosomal maturation. Other species prevent acidification of the phagosome or nullify ROIs and RNIs. Escape from the phagosome into the host cytosol is a strategy used by pathogenic bacteria and can occur at multiple stages of phagosome maturation. Certain bacteria exploit PRRs to enable self-replication in modified endosomes or modulate iron (Fe) abundance. Inhibition of phagolysosome formation, apoptosis, and autophagy also contribute to establishment of infection with intracellular bacteria. DAMP, danger-associated molecular pattern; NADPH, nicotinamide adenine dinucleotide phosphate-oxidase.
Invasion of Nonprofessional Phagocytes
Microbe-directed uptake allows entry into nonphagocytic cells and hence can be seen as an evasion mechanism of phagocytosis by professional phagocytes. The target spectrum of intracellular bacteria ranges from very broad to highly specific. M. leprae is found in a large variety of host cells and hence shows a broad target cell spectrum. L. monocytogenes enters the host through the gut epithelium and its major target besides MPs is the hepatocyte; M. tuberculosis is almost, if not exclusively, restricted to MPs, although pneumocytes have been proposed as a safe niche in the lung. It is noteworthy that intracellular bacteria are often capable of entering a variety of cell lines in vitro. These in vitro experiments do not necessarily reflect an in vivo situation, and care should be taken in extrapolating conclusions from them. For obligate intracellular bacteria, nonprofessional phagocytes rather than MPs represent the preferred habitat. These bacteria are primarily found in endothelial and epithelial cells.17,21
Recognition and Downstream Events
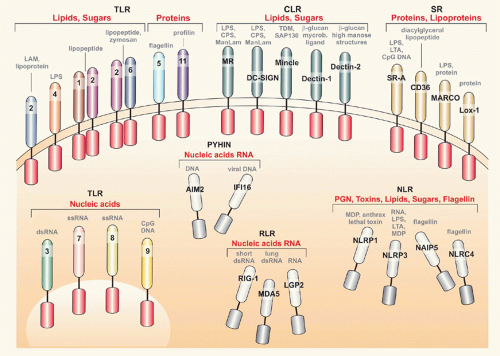
Microbes are composed of various molecules that are structurally different from host cell/tissue composition (ie, lipopolysaccharide [LPS], peptidoglycan [PGN], cytosinphosphatidyl-guanosin [CpG] deoxyribonucleic acid [DNA]). These microbe-specific molecules encompass various biochemical entities and have been named pathogenassociated molecular patterns (PAMPs).45 Once pathogens assault host tissues, PAMPs are recognized by pattern recognition receptors (PRRs). PRRs are nonclonally distributed on various cell types; they are germline encoded and their activation, following PAMP ligation, is an essential event for initiation of the immune response and disposal of the intruder.46,47,48,49 Microbial ligand diversity parallels the receptor repertoire, and currently there are several classes of PRRs known (Fig. 40.2), which may be classified based on chemical structure as detailed in the following:
Toll-like receptors (TLRs) are type-I transmembrane proteins. Currently, 10 TLR members in humans and 12 in mice have been described.50 TLRs 1, 2, 4, 5, and 6 are cell surface-associated, and TLRs 3, 7, 8, and 9 are associated with vesicles of the endoplasmic reticulum (ER), endosomes, and lysosomes (see Fig. 40.2).51 TLRs are signaling receptors and recruit single or a combination of toll-interleukin (IL)-1 receptor-resistance (TIR)-containing adaptors, including myeloid differentiation primary response gene 88 (MyD88), TIR domain-conining adaptor protein, TIR domain-containing adaptor-inducing interferon-beta (IFN-β, and TIR domain-containing adaptor-inducing IFN-β-related adaptor molecules.46 Timing (sequential versus simultaneously) and space (vacuole characteristics, early versus late phagosome) are essential for TLR downstream effects. TLR-4 senses LPS and thus detects extracellular gram-negative bacteria. TLR-4 first initiates nuclear factor kappa-beta (NF-κB) and secondly IFN I pathway activation via MyD88 and TIR domain-containing adaptor-inducing IFN-β, respectively. Once bacteria are phagocytosed, TLR-9 senses CpG DNA and signals via MyD88 to NF-κB in early endosomes while in lysosomes Traf3/IRF3 is assembled to enable IFN I responses.50 TLR activation may impact the microbicidal capacity of the infected cell.52
C-type lectin receptors (CLRs) belong to a large superfamily of membrane or soluble proteins, which have one or more calcium-dependent carbohydrate-binding lectin domains (see Fig. 40.2).53,54 CLRs have high avidity for carbohydrates/glycans. CLRs mediate endocytic uptake, a feature distinct from that of TLRs. Signaling is mediated by immune-receptor tyrosine-based activation motif/spleen tyrosine kinase (Dectins; macrophage-inducible C-type lectin, Mincle), hemITAM (Dectin-1), immune-receptor tyrosine-based inhibition motif, and the kinase Raf1 (Dectin-1, dendritic cell [DC]-specific intercellular adhesion molecule-3-grabbing nonintegrin [SIGN]) to activate mitogenactivated protein kinase (MAPK) and NF-κB. CLRs are involved in generation of reactive oxygen species via the syk pathway in myeloid cells, modulate adaptive immune responses, and seem to be indispensable elements for T helper (Th)17 responses. Other CLR, notably DC-SIGN, induce inhibitory signals.
Nucleotide oligomerization domain (NOD)-like receptors (NLRs) are cytosolic proteins (see Fig. 40.2). This family comprises more than 20 members, which are structurally diverse and are involved in pathogen sensing (NOD1, NOD2) and initiation of the inflammatory response (nucleotide-binding domain and leucine-rich repeat with pyrin domain-containing [NLRP]1, NLRP3, nucleotide-binding domain with leucine rich repeat and caspase recruitment domain [NLRC]4; NLRC5).55,56,57 NOD1 and 2 use the receptorinteracting protein 2 adaptor to signal and are involved in PGN recognition. Thus, they are essential for sensing cytosolic bacteria in professional and nonprofessional phagocytes. NRLP and NLRC members, as well as NOD2, are essential components of inflammasomes, which are cytosolic platforms responsible for secretion of IL-1β, as detailed later.
Retinoic acid-inducible gene I (RIG-I)-like receptors (RLRs) are cytosolic ribonucleic acid helicases (RIG-I, MDA5, and LGP2) involved in antiviral immunity.58
Scavenger receptors (SRs) are cell membrane proteins that recognize modified host molecules, as well as microbial structures.59 These receptors are mainly involved in uptake of particles including intracellular bacteria (eg, macrophage receptor with collagenous structure, MARCO; SR-A, cluster of differentiation [CD]36) (see Fig. 40.2).
Pyrin- and HIN-containing proteins (PYHINs) have been recently associated with pathogen sensing (see Fig. 40.2). Absent in melanoma 2 (AIM2) and gamma-IFN-inducible protein 16 are the most prominent receptors involved in DNA sensing and consequently modulation of IFN secretion and inflammation.60 AIM2 is associated with inflammasome activation and senses host and bacterial cytosolic DNA.
A key issue for host defense is the receptor profile at particular time and tissue site of infection. PRR expression in phagocytes is influenced by cell type (resident tissue macrophage versus inflammatory monocytes), tissue (lung versus gut), milieu, and activation status (certain cytokines up- or downregulate receptors).61 Importantly, multiple PAMPs engage multiple PRRs simultaneously or sequentially and therefore, a given pathogen by means of specific combinations of PAMPs can tailor the host response in a highly specific way. TLRs can form dimers (eg, TRL-1/2 or TLR-2/6), and may collaborate with distinct CLRs (TLR-2/Dectin-1; TLR-2/MARCO; TLR-4/CD14). On the other hand, a given PAMP may be sensed by multiple PRRs (glucans sensed by Dectin-1; complement receptor [CR]; CD36). Thus, PRRs orchestrate pathogen- and cell type-specific host immune responses. Pathogens able to alter molecules or modulate host cell death add an additional layer to this recognition process by inducing activation of PRRs via recognition of pathogens in combination with sensing of danger signals. Certain PRRs are involved in sensing both microbial components and danger signals. For instance, Mincle recognizes mycobacterial trehalose dimycolate (TDM) and the ribonucleoprotein spliceosome-associated protein 130, a protein associated with eukaryotic cell death. Despite their name, PAMPs are not restricted to pathogens but are also expressed by nonpathogenic microbes. Hence, the host probably needs to sense pathogen-specific signals in addition. In the case of intracellular bacteria, such signals could emanate from bacterial persistence, that is, from duration of PAMP expression. More recently, bacterial messenger ribonucleic acid was suggested as a signal from viable bacteria (vitaPAMP), which alerts the immune system.62
FIG. 40.2. The Pattern Recognition Receptors. Bacterial recognition is paramount in the crosstalk between host and pathogen. Several classes of receptors contribute to this process. Toll-like receptors are present at the surface membrane or within ensodomal compartments and recognize lipids, carbohydrates or proteins. C-type lectin receptors and scavenger receptors are expressed at the cell surface and sense glycolipids and lipoproteins, respectively. Surveillance of the cytosol is mainly performed by nucleotide oligomerization domain-like receptors (NLRs), RIG I-helicase receptors (RLRs), and pyrin- and HIN-containing proteins (PYHINs). RLRs are exclusively involved in viral detection, whereas NLRs recognize a wide range of structures. PYHIN detect deoxyribonucleic acid from various sources. AIM2, absent in melanoma 2; CpG, cytosinphosphatidyl-guanosin; CPS, capsular polysaccharide; DC-SIGN, dendritic cell-specific intercellular adhesion molecule-3-grabbing nonintegrin; ds, double stranded; FI16, gamma-interferon-inducible protein 16; LAM, lipoarabinomannan; LGP2, laboratory of genetics and physiology 2; Lox-1, lectin-type oxidized LDL receptor 1; LPS, lipopolysaccharide; LTA, lipoteichoic acid; ManLAM, mannosylated lipoarabinomannan; MARCO, macrophage receptor with collagenous structure; MDA5, melanoma differentiation-associated gene 5; MDP, muramyl dipeptide; Mincle, macrophage-inducible C-type lectin; MR, mannose receptor; NAIP, baculoviral IAP repeat-containing protein; NLRP, NACHT, LRR and PYD domaincontaining protein; NLRC, NLR family CARD domain-containing protein 4; PGN, peptidoglycan; RIG-I, retinoic acid-inducible gene I; ss, singlestranded; TDM, trehalosedimycolate; SAP130, spliceosome-associated protein 130.
PRRs may be classified according to pathogen-binding propensity and impact on uptake as:
Opsonic receptors: Fc receptors (FcRs), CRs, pentraxins, surfactant proteins, ficollins
Nonopsonic receptors: TLRs, NLRs, RLRs
Based on their effects on pathogen internalization, PRRs may be classified as:
Phagocytic receptors: CRs, FcRs, mannose receptors (MRs), and DC-SIGN (CLRs), CD36, MARCO, and SR-A (SRs)
Signaling receptors: TLRs, NLRs, RLRs, CLRs
Bacterial cell wall composition, secretion products, as well as intracellular location are fundamental aspects for recognition of intracellular bacteria. M. tuberculosis is rich in several classes of PAMPs. This pathogen interferes with the endocytic pathway and resides in an early phagosome. Phagocytosis is mediated by CLRs (MRs, DC-SIGN) and SRs (CD36), which primarily recognize mannosylated cell wall mannans and by opsonic receptors, FcRs, and CRs. Another CLR, namely Langerin, seems to have a role in M. leprae recognition. Lung surfactant proteins mediate the recognition of tubercle bacilli in the alveolar space. Sensing of the lipopeptides and phosphatidylinositol mannans is performed by TLR-2, while TLR-4 involvement is still debated. Multiple CLRs sense mycobacterial glycans or lipoglycans. TDM, a specific component of mycobacteria, is recognized by Mincle, while DC-SIGN senses mannosylated lipoarabinomannan. SIGNR3 and Dectin-1 and -2 are also involved in recognition. However, to date no specific structure from mycobacteria has been identified as a ligand for these lectins. Once in the phagosome, bacteria may be sensed by TLR-9. Bacterial PGN, which accesses the cytosol, is recognized by NOD2. Notably, there is a degree of redundancy between receptors belonging to different classes, as revealed by studies with mice with single or multiple deficiencies in TLRs, NLRs, and SRs.63 It appears that adaptors able to gather signals from various PRRs, such as MyD8864 and caspase recruitment domain-containing protein 9 (Card9)65 are key in controlling susceptibility to TB.
Recognition of Salmonella, which lives in a late phagosome, is dominated by TLR members. LPS and the lipopeptides from the cell wall are sensed by TLR-2 and -4 before internalization. Ablation of these TLR renders mice highly susceptible to disease.66 Vacuolar bacteria are recognized by TLR-9 via CpG DNA. Interestingly, triple-deficient (TLR- 2, -4, -9) animals are resistant as simultaneous TLR activation is necessary for acidification of the vacuole, which in turn induces Salmonella pathogenicity island 2 (SPI2) genes. Virulence effectors are translocated into the cytosol transforming the phagosome into a replicative niche for Salmonella.67 Flagella are sensed by TLR-5 and NLRC4.
Recognition of L. monocytogenes, which escapes from phagosome into cytosol, is complex. Cell wall lipopeptides are sensed by TLR-2, while PGN is recognized by NOD1 and NOD2. MyD88 seems to be essential for defense against listeriae.68 Bacterial DNA is monitored by multiple sensors in the cytosol, including AIM269,70,71,72,73 and leucine-rich repeat flightless-interacting protein 1.74Francisella, which also egresses into the cytosol, is recognized in a similar way by multiple receptors, including AIM270,75 and NLRC4.
By sensing the broad molecular spectrum of pathogens, PRRs induce cellular programs as the first line of defense, including antimicrobial effector functions and maturation of DCs for instruction of the adaptive immune response. However, PRRs and their adaptors may be exploited by intracellular pathogens to escape killing and establish stable infection. Mycobacteria use CRs to ensure a safe entry into macrophages and limit maturation of the phagosome. They also use the TLR/MyD88 pathway to induce cytokines (IL-6 and IL-10), which through signal transducer and activator of transcription (STAT)3 signalling bias arginine metabolism to arginase-1 over nitric oxide production by inducible nitric oxide synthase (iNOS).76,77 In addition, polymorphonuclear leukocyte encounters with mycobacteria results in MyD88/Card9-dependent IL-10 release to dampen immune responses.65,78 Similarly Yersinia uses TLR-2 to induce IL-10.79
Phagocytosis and Phagosome Dynamics
Phagocytosis of inert particles initiates a series of events that ultimately lead to the formation of a phagolysosome (see Fig. 40.1).80,81 Phagosome maturation is a strictly coordinated sequence of fusion and fission events, which involves defined compartments of the endocytic pathway.81,82 Immediately after or even during phagosome sealing, phagosome maturation proceeds.
The early phagosome is characterized by a slightly acidic to neutral pH (6.0 to 6.5) and membrane markers, such as MR, the tryptophane aspartate-containing coat protein, and the transferrin receptor (TfR) with its ligand transferrin, small GTPase (Rab5), early endosomal antigen 1, phosphatidylinositol 3-phosphate, and Syntaxin13.
The late phagosome is characterized by a pH between 5.0 and 6.0 and the acquisition of the vacuolar adenosine triphosphatase (ATPase) proton pump (V-H+ATPase), mannose-6 phosphate receptor, Rab-interacting lysosomal protein, and Rab7.
The phagolysosome results from the fusion between phagosomes and lysosomes, characterized by a pH between 4.0 and 5.5, high density of lysosome-associated membrane proteins, and typical lysosomal enzymes (such as cathepsins).
The three stages form a continuum involving the sorting of membrane proteins, as well as budding of, and fusion with, other vesicles. During this dynamic process, the phagosomes successively interact with the corresponding endosomes and subsequently with lysosomes.81 Characterization of the Rab family of GTPases on vacuoles harboring pathogens facilitates identification of the host membrane transport pathways, which are turned on during infection.83M. tuberculosis and S. enterica interfere with the endocytic pathway by retaining Rab5 and Rab7, respectively, on their vacuoles.84L. pneumophila, Brucella abortus, and C. trachomatis interact with the secretory pathway as revealed by the presence of Rab1, Rab2, and Rab6, respectively, on their phagosomes.85,86
Acquisition of a vacuolar ATPase proton pump plays a central role in acidification of the phagosome.81,87 Immediately after phagocytosis, the phagosome becomes alkaline for a short time before acidification is initiated. The basic milieu is optimal for the activity of defensins and basic proteins, whereas the acidic pH is optimal for lysosomal enzymes. Defensins are small (3.5 to 4.0 kD) peptides rich in arginine and cysteine.88,89 They are abundant in PNG and present in some, though not all, MPs (depending on species and tissue location). Purified defensins are microbicidal for certain intracellular bacteria, such as S. enterica and L. monocytogenes. The contribution of lysosomal enzymes to bacterial killing is likely minor. Their major task is the degradation of already killed bacteria. These enzymes reside in the lysosome and are delivered into the phagosome during maturation through several independent waves, and they reach their optimum activity during later stages, that is, in the phagolysosome.
Most intracellular bacteria interfere with phagosome maturation and alter the phagosome in order to facilitate their own survival (see Fig. 40.1).80,90,91 These include L. pneumophila, M. tuberculosis, S. enterica, C. burnetii, and Chlamydia. Although the specific mechanisms are incompletely understood, mycobacterial sulfatides and some mycobacterial glycolipids, most notably mannosylated lipoarabinomannans, impede phagolysosome fusion. Mycobacterial products, such as SapM and MptpB, contribute as well to the arrest of maturation of the early endosome. Antibody-coated M. tuberculosis organisms lose their capacity to block discharge of lysosomal enzymes, suggesting an auxiliary function of antibodies in cell-mediated protection against TB.92 Finally, the robust, lipid-rich cell wall of mycobacteria renders them highly resistant against enzymatic attack. M. tuberculosis, as well as M. avium, arrest phagosome maturation at an early stage. They restrict phagosome acidification via the exclusion of the V-H+ATPase proton pump from the phagosome. Additional mechanisms may contribute to this event, such as NH4+ production by M. tuberculosis. Consistent with intraphagosomal NH4+ production, the urease of M. tuberculosis is active at low pH. It has been known for some time that NH4+ also interferes with phagosome-lysosome fusion. Exogenous adenosine triphosphate (ATP) has been shown to promote phagolysosome fusion resulting in concomitant death of macrophages and killing of M. bovis bacillus Calmette-Guérin (BCG).93,94
Phagosome maturation is arrested somewhere between the early and late stages by M. tuberculosis, M. bovis BCG, L. pneumophila, S. enterica, and C. trachomatis, all of which replicate in nonacidified vacuoles. Phagosomes containing S. enterica, M. bovis BCG, or C. trachomatis appear uncoupled from the maturation process through which phagosomes containing inert particles proceed.80,90S. enterica remains in the spacious membrane-bound phagosome, which is formed after uptake by the trigger mechanism. Moreover, the bacteria manipulate the cytoskeleton via kinesin and tether the vacuole to membranes of the Golgi apparatus.95 The vacuole containing C. trachomatis, which lacks any specific phagosome markers, is loaded with ATP, which is required by the pathogen, by an unknown mechanism. In addition, elements of the cytoskeleton (actin and filamin) are used to stabilize the vacuole.96L. pneumophila prevents fusion of the vacuole with the endosomal compartment and recruits vesicles derived from Golgi and ER97 by use of the T4SS. Moreover, Chlamydia employ mimics of soluble NSF attachment protein receptor to modulate membrane transport. Legionella uses an array of proteins to control Rab1 activity in the vacuole.98,99 Bacterial DrrA (SidM protein) is a highly efficient guanine nucleotide exchange factor for Rab1A.44C. burnetii have evolved to live in the acidic late phagosome.82,100 Recent in vitro studies confirmed Coxiella‘s requirement for low pH and oxygen tension. The fusogenic properties of the vacuole are tightly regulated by the T4SS apparatus, through ankirin proteins.101
Egression into the Cytoplasm
Egress from the phagosomal into the cytoplasmic compartment represents a highly successful microbial survival strategy because bacterial killing is focused on the phagolysosome to limit self-damage of MPs. This egression has been extensively studied in L. monocytogenes, but is known to be utilized by other intracellular pathogens, including shigellae, rickettsiae, francisellae, and mycobacteria (see Fig. 40.1).102
Cytoplasmic invasion by L. monocytogenes depends on listeriolysin (LLO), an SH-activated cholesterol-dependent cytolysin. LLO requires activation by a host factor (gamma-IFN-inducible lysosomal thiol reductase), a thiol reductase.103 In the cytosol, LLO is degraded, thus avoiding killing of host cells.104 Deletion of the LLO gene (hly) renders L. monocytogenes avirulent. LLO is also required for replication of L. monocytogenes in spacious listeria-containing phagosomes. These compartments are nonacidic and allow slow replication of the pathogen.105 Other molecules, such as phospholipase and lecithinase, are likely involved in membrane transition but are insufficient on their own. Invasion of L. monocytogenes into the cytoplasmic compartment is markedly reduced in IFNγ-activated macrophages in which the microbe, entrapped in the phagosome, rapidly succumbs to attack by toxic oxygen and nitrogen species and/or defensins. Cytosol evasion of shigellae is mediated by factors, which are also involved in bacterial entry (eg, IpaB, product of T3SS). Listeria simultaneously activates caspase-1 and consequently modulates death of infected cells via danger signals represented by remnant vacuolar membranes.106M. tuberculosis uses proteins encoded by the T7SS, located in the region of difference 1 gene region, to successfully translocate to the cytosol.5,107
Cell-to-Cell Spreading
L. monocytogenes is cleared from the blood by Kupffer cells and then spreads to adjacent hepatocytes without reentering the extracellular milieu. This mechanism of cell-to-cell spreading has been carefully studied in vitro.108 Having entered the cytoplasm, L. monocytogenes induces a tail of host actin filaments, which push the bacterium forward to the outer region of the cell, where it induces pseudopod formation. Intracellular movement is achieved by coordinated actin polymerization at, and polarized release from, the bacterial surface. The ActA gene encodes a 90-kDa protein located on the bacterial surface, which is responsible for these actin-based movements.109 A host cytosolic complex composed of eight polypeptides has been identified which, on binding Act A, induces actin polymerization.110 The pseudopod-containing L. monocytogenes is engulfed by the adjacent cell, and the microbe reaches the phagosome of the recipient cell, which is still enclosed by cytoskeletal material from the donor cell. The two plasma membranes of the host and recipient cells apparently fuse, thereby allowing the introduction of L. monocytogenes into the cytoplasm of the recipient cell. Thus, L. monocytogenes can infect numerous cells without contacting extracellular defense mechanisms. Shigella use similar mechanisms for evasion and intracellular movement, and a similar spreading mechanism seems to be employed by S. enterica and by R. rickettsii, but not by R. prowazekii and R. typhi. A role for motility and manipulation of host actin-based structures was recently demonstrated for virulent mycobacteria.111 Bacterial T7SS mediates ejection from the infected host cell and facilitates spreading through actin structures coined “ejectosomes.”
Cell Death Patterns
Death of mammalian cells occurs by accidental or programmed cell death, which were once thought to be associated exclusively with necrosis or apoptosis, respectively.112 Increasing evidence, however, suggests that necrosis can progress in a programmed sequence. Moreover, in addition to necrosis and apoptosis, additional modes of cell death have been described recently, namely autophagic cell death, pyroptosis, pyronecrosis, necroptosis, mitotic catastrophe, NETosis, and lysosomal membrane permeability.113,114,115 Although autophagy is a complex process aimed at preventing cell death, it has emerged as relevant mechanism to control infection and hence will be described in this section. Generally, intracellular pathogens often counteract host cell death in order to maintain their habitat. Thus, ability of microbes to modulate eukaryotic cell survival evolved as an essential pathogenicity feature.
Apoptosis and Necrosis
Apoptosis is a tightly controlled process that is initiated by intrinsic mechanisms within the dying cell. It involves a series of tightly controlled enzymatic events, notably intracellular caspases. Necrosis is the result of cell destruction caused by various exogenous effector mechanisms, including those mediated by complement and cytolytic T-lymphocytes. In contrast to necrosis, apoptosis is generally noninflammatory and thus associated with tissue repair rather than destruction. However, cell death associated with bacterial infection frequently results in release of microbial PAMPs, which serve as inflammatory signals. Intracellular bacteria interfere with apoptosis in various ways to delay or even block this process and thus sustain their preferred habitat. C. burnetii antagonizes the intrinsic apoptotic death of macrophages by means of ankirin proteins related to T4SS.116,117A. phagocytophilum blocks PNG apoptosis by secreting the Ats1 protein via T4SS across the phagosomal membrane. Ats1 enters the mitochondria and subverts apoptotic signaling.118Chlamydia prevents immature cell death and uses autophagy in addition.113L. pneumophila induces rapid apoptosis in DCs,119 but prevents programmed cell death in other cell types.120 Moreover, this pathogen limits multiple pathways leading to cell death, including necrosis in macrophages.121 Mycobacteria offer a good example of how virulence is associated with modulation of death of infected cells. Avirulent mycobacteria induce apoptosis, while M. tuberculosis preferentially causes necrosis.122 Tubercle bacilli interfere with eicosanoid-regulated cell death to facilitate necrosis. Generally, death of infected cells impacts on the acquired immune responses, pathology, and ultimately disease manifestation.
Autophagy
Autophagy is a catabolic process that controls the integrity of eukaryotic cells.123 This mode of “self-digestion” is paramount for the disposal of protein complexes that cannot be degraded via the proteasomal route and for the elimination of damaged organelles. Three different autophagic processes have been described: chaperone-mediated autophagy, microautophagy, and macroautophagy (canonical autophagy).115 A unique feature of autophagy is the direct sequestration of the cargo into autophagosomes, which are surrounded by a double membrane and delivered to the lysosomal compartment. Autophagy is tightly orchestrated by over 30 autophagic components. This cell-autonomous housekeeping process is also an efficient system for the elimination of intracellular pathogens. Direct autophagy of intracellular pathogens occurs in any cell type and has been named xenophagy.124 As a process of cytosol surveillance, autophagy is directed primarily against pathogens that egress into the cytosol. However, most bacteria, which successfully adapted to the intracellular milieu, have developed mechanisms to protect themselves against autophagy. Some species even harness the autophagic machinery to their advantage.
Autophagosome-like structures were first described in PNG infected with Rickettsia,125 and a role for autophagy in control of intracellular bacteria was first demonstrated for M. tuberculosis.126 Subsequently, the protective functions of autophagy were established in infections with M. tuberculosis,127,128S. enterica,129 and L. monocytogenes.130,131,132 Microbe-derived PAMPs stimulate autophagy131,133 as do cytokines like IFNγ, most likely through induction of GTP/guanylate-binding protein (GBP) molecules.134,135 Lipids were also found critical for autophagy in intracellular bacterial infections. Thus, the signaling lipid diacylglycerol is required for autophagy induced by S. typhimurium.136 Recently the term sequestosome-like receptors was coined to define cytosolic innate receptors (p62, NBR1, NDP52), which target intracellular pathogens to the autophagic machinery115 including salmonellae,137,138 shigellae,106 listeriae,139 and mycobacteria.140
Pathogens interfere with autophagy through multiple strategies: by blocking induction or maturation of autophagosomes into autolysosomes, by evading recognition by the autophagic machinery, and by misusing autophagy for their own benefit. Microarray studies revealed that F. tularensis downregulates autophagy-related genes.141 Similarly, whole genome-wide profiling of macrophages infected with M. tuberculosis suggests that host factors that regulate bacterial replication are regulators of autophagy.142S. enterica blocks autophagy by interference with the ubiquitination machinery,137,138 and L. pneumophila induces autophagy, but delays fusion with lysosomes.143 A similar scenario was reported for C. burnetii144 and A. phagocytophilum.145 A different strategy is employed by bacteria that egress into the cytosol. The shigella protein VirG targets bacteria to the autophagic pathway by binding to the autophagic protein Atg5. However, once in the cytosol, the pathogen uses the T3SS IcsB to competitively bind to Atg5 and thus renders it unavailable for VirG.146 Similarly, ActA impedes autophagic targeting of listeriae by recruiting actin and protecting it.139,147 Some intracellular bacteria not only affect autophagy but may also exploit it. Thus, chlamydiae gain access to nutrients by stimulating autophagy,148 and listeriae exploit this process for establishing chronic infection.105F. tularensis reenters the endocytic pathway following its egression into the cytosol through autophagosomes.149 Autophagy is implicated in the biogenesis of vacuoles and persistence of C. burnetii therein.150
Pyroptosis
Pyroptosis is a form of programmed cell death that is controlled by caspase-1. It is an inflammatory process that results in rapid lysis of infected cells, mostly macrophages.114 This process was first described for shigellae151 and subsequently for salmonellae,152,153 legionellae, and burkholderiae.154 Mouse studies showed that pyroptosis is an innate immune effector mechanism during infection with S. typhimurium.154 It still needs to be clarified how intracellular bacteria escape or modulate pyroptosis.
Intracellular Iron
Iron is required by most organisms and is a cofactor for enzymes involved in many essential biologic processes. The same divalent cation is toxic at high concentration and therefore is tightly controlled by multiple elements.155 As both host and intracellular pathogens require iron, microbes have evolved ways to interfere with iron homeostasis. Generally, the strategy of the mammalian host is to deprive the pathogen of iron. However, depending on microbial habitat (extracellular versus intracellular bacteria), a given mechanism may be beneficial or detrimental. While systemic iron withdrawal and hypoferremia contribute to the control of extracellular bacteria, these strategies are counterproductive for intracellular microbes. Rather, down regulation of iron uptake and augmentation of iron export are advantageous for the host in defense against intracellular pathogens.156,157
Intracellular bacteria require iron, and production of reactive oxygen intermediates (ROIs) and reactive nitrogen intermediates (RNIs) also depends on iron. Thus, competition for the intracellular iron pool between the intracellular pathogen and the host cell markedly influences the outcome of their relationship.158 To improve their iron supply, mammalian cells utilize specific molecules. In the extracellular host milieu, iron is tightly bound to transferrin and lactoferrin, and the transferrin-iron complex is taken up by host cells via TfRs. The lactoferrin-iron complex does not enter the cell. Iron is released from the transferrin-TfR complex under the reducing conditions of the early phagosome. This event is controlled by Hfe (the product of the hereditary hemachromatosis gene).155 Hfe reduces iron uptake either by inhibiting TfR internalization or by blocking iron release from transferrin in the early phagosome. Macrophages can also acquire iron via the divalent metal transporter-1. Iron may also bind to haptoglobin, haemopexin, and lipocalin-1 and -2 in the extracellular space. Within the endocytic pathway, the Nramp system is involved in iron transport from the phagosome to the cytosol, where iron is bound to ferritin. Ferroportin (FPN) is one of the best-characterized iron export molecules in eukaryotic cells. Hepcidin, a product not only of liver cells but also macrophages, regulates FPN activity by inducing FPN internalization and proteasomal degradation. Accordingly, iron availability is controlled in multiple ways, including lactoferrin concentration in the extracellular space, intracytosolic ferritin concentration, hepcidin levels and abundance of TfRs, divalent metal transporter-1, and FPN on the cell surface. Many intracellular bacteria, including M. tuberculosis, L. pneumophila, and S. enterica, accommodate themselves in the early phagosome, where the abundance of iron-loaded transferrin guarantees a high availability of iron. Moreover, these bacteria, as well as Chlamydia and F. tularensis, induce divalent metal transporter-1 expression to support their replication.159 Hepcidin is induced in mycobacterial phagosomes160 and limits iron export by degrading FPN. Hepcidin also promotes Salmonella‘s growth.161 Similar observations, suggesting that iron efflux is detrimental for intracellular pathogens, were reported for C. psittaci and L. pneumophila.162 To successfully compete for iron, bacteria possess a variety of iron-binding proteins. These include iron chelators (siderophores), transferrin-binding proteins, hemelike proteins, and ATP-binding cassette transporters.163,164,165 Expression of genes involved in iron uptake is controlled by a conserved mechanism involving the Fur protein. However, lipocalins, which are host proteins produced by professional phagocytes and epithelial cells, bind siderophores from mycobacteria and salmonellae and thus minimize iron usage by microbes. Moreover, deletion of lipocalin-2 impacts on invasion of epithelial cells by tubercle bacilli.161,166
Importantly, there are many crossregulatory interactions linking iron homeostasis and immune responses. Th1 immunity and iron availability are interconnected. IFNγ-activated MPs downmodulate TfR expression and intracellular ferritin, resulting in reduced iron availability within the phagosome. Overall, IFNγ induces iron sequestration by downregulating FPN, while IL-4 favors iron release.156,167 IFNγ, on the other hand, induces Nramp1, whose relevance for protection against salmonellae and mycobacteria has been established.168 The iron content of the phagosome in the resting MP seems to be sufficient for L. pneumophila. However, available iron is markedly reduced in IFNγ-activated MPs and, as a consequence, L. pneumophila, which lacks efficient iron uptake mechanisms, starves from iron deprivation in activated macrophages. In contrast, M. tuberculosis possesses a potent iron acquisition system comprising exochelins and mycobactins. The exochelins successfully compete for iron under limiting conditions and transfer it to mycobactins in the cell wall.169,170 Similarly, SPI2 are upregulated as a response to Nramp-1-induced iron starvation and modify the vacuole to ensure bacterial replication.
Toxic Effector Molecules
Killing of intracellular bacteria by MPs and/or PNGs is primarily accomplished by highly reactive toxic molecules, notably ROIs and RNIs.171,172,173,174 Within infected cells, these molecules are bactericidal. However, both ROIs and RNIs have a broader functional spectrum. They are also involved in signaling, regulation of vascular tone, tissue injury, and control of inflammation. Thus, ROIs and RNIs can both alleviate and promote tissue damage.171
Most if not all bacteria are susceptible to ROIs in vitro. Yet, contribution of ROIs to killing of intracellular bacteria by MPs is less clear; in murine macrophages, RNIs are more important. On the contrary, in PNGs, the role of ROIs seems to prevail.175 In the mouse, ROIs and RNIs act consecutively in defence against S. enterica infection.176,177 Production of RNIs by human MPs at concentrations sufficiently high for bacterial killing remains controversial.178,179,180 However, evidence is accumulating that human MPs from sites of intracellular bacterial infection produce adequate concentrations of RNIs. Thus, using antibodies with exquisite specificity for human iNOS, this enzyme could be detected in a large proportion of lung macrophages from patients with TB.181,182,183,184 iNOS can be induced by different mechanisms in different species as suggested, for example, by divergence of the iNOS promoter in mouse and human.171,185,186
ROI production is initiated by a membrane-bound nicotinamide adenine dinucleotide phosphate-oxidase (NADPH oxidase) complex composed of six enzyme units (Rho guanosine triphosphatase, gp91PHOX also named NOX2, p22phox, p40phox, p47phox, and p67phox). NADPH oxidase is activated by IFNγ and by immunoglobulin (Ig)G-FcR binding:
O2– is further metabolized by superoxide dismutase:
In the presence of appropriate iron catalysts, the Haber-Weiss reaction takes place:
In addition, O2– is transformed into1O2. The1O2 and•OH radicals are short-lived powerful oxidants with high bactericidal activity causing damage to DNA, membrane lipids, and proteins. (Note: O2–, hyperoxide anion;*OH, hydroxyl radical containing a free electron;1O2, singlet oxygen, a highly reactive form of O2.)
Granulocytes, blood monocytes, and certain populations of tissue macrophages possess myeloperoxidase, thus allowing halogenation of microbial proteins172:
In addition to hypochlorous acid, chloramines are formed and both agents further increase the bactericidal power of the ROI system by destroying biologically important proteins through chlorination.
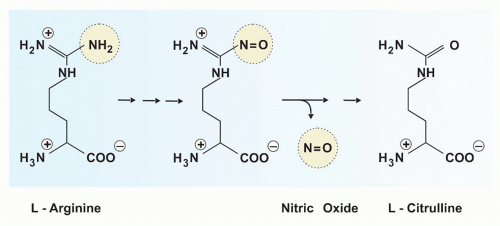
Nitric oxide is exclusively derived from the terminal guanidino nitrogen atom of L-arginine (Fig. 40.3). This reaction is catalyzed by the iNOS, which leads to the formation of L-citrulline and NO•.
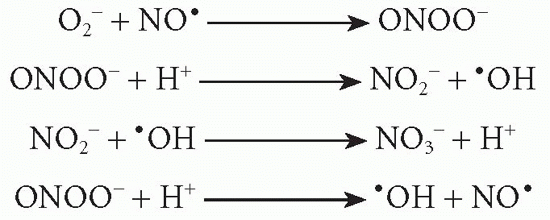
NO• can act as oxidizing agent alone or interact with O2– to form the unstable peroxynitrite (ONOO–). This may then be transformed to the more stable anions, NO2– and NO3–, or decomposed to NO•:
NO• and ONOO– are highly reactive antimicrobial agents. NO• may be transformed to nitrosothiol, which expresses the most potent antimicrobial activity. In contrast, NO2– and NO3– are without notable effects on microorganisms.
Three distinct nitric oxide synthase isoenzymes are known. The two constitutive nitric oxide synthases (neuronal nitric oxide synthase and endothelial nitric oxide synthase) exist in various host cells and account for basal nitric oxide synthesis, whereas iNOS is primarily found in professional phagocytes and is responsible for microbial killing. Its induction is controlled by exogenous stimuli such as IFNγ, agonist of PRRs, and inflammatory cytokines. This iNOS stimulation results in a burst of high RNI concentrations required for microbial killing, whereas the low nitric oxide levels produced by neuronal nitric oxide synthase and endothelial nitric oxide synthase perform physiologic functions. The RNIs exert their bactericidal activities by directly inactivating iron-sulfur-containing enzymes, by S-nitrosylating proteins, by damaging DNA, or by synergizing with ROIs. Both ROIs and RNIs are relevant for killing of Salmonella, while most other intracellular pathogens are primarily controlled by iNOS products. In chlamydial infections, insufficient as well as excessive production of nitric oxide can be immunosuppressive.187 RNIs prevail as antimicrobial molecules against L. monocytogenes,188C. burnetii,189C. trachomatis, F. tularensis, Brucella, and L. pneumophila.190 The pivotal role of iNOS in control of M. tuberculosis infection has been demonstrated in the mouse system.191 Despite resistance of NOX2-ablated mice against TB, recent data supporting a role of macrophage ROIs in humans have been reported.192 Thus, nitrosative and oxidative processes under the control of iNOS and NADPH oxidase together are paramount for protection against intracellular bacteria.
FIG. 40.3. Generation of Nitric Oxide from L-Arginine. L-Arginine is metabolized to the toxic molecules nitric oxide and L-citrulline by inducible nitric oxide synthase.
Evasion of Killing by Reactive Oxygen Intermediates and Reactive Nitrogen Intermediates
Microbes use multiple strategies to nullify ROI and RNI attack: evasion, inhibition, enzymatic inactivation, generation of scavenger molecules, as well as stress and repair mechanisms (see Fig. 40.1).171 SPI2 enables S. enterica to exclude NADPH oxidase from the phagosomal membrane, thus interfering with ROI release into the Salmonella phagosome. S. enterica mutants deficient in SPI2 are susceptible to ROIs.193 Similarly, proteins translocated into the cytosol interfere with iNOS activity.194F. tularensis undergoes phase variation to switch to a phenotype that is less stimulatory for iNOS.195A. phagocytophilum inhibits ROI production in PNGs after an initial ROI induction process.196 Many intracellular bacteria produce superoxide dismutase and superoxide catalase that detoxify O2 and H2O2, respectively.171,197 Production of ROI-detoxifying molecules by intracellular bacteria is not constitutive; rather, expression of these enzymes is controlled by regulators such as soxR or oxyR that sense for concentrations of O2 or H2O2, respectively. Accordingly, transposon mutants of S. enterica that fail to survive inside murine MPs are highly sensitive to ROI in vitro. Although less is known about specific mechanisms by which intracellular bacteria interfere with killing by RNIs, catalase and other antioxidative enzymes may indirectly inhibit RNI functions. ROI- and RNI-detoxifying gene products have been identified in M. tuberculosis.198 KatG,199 SodA, and SodC are involved in these processes. Recently, mycobacterial nicotinamide adenine dinucleotide dehydrogenase and enhanced intracellular survival (eis) gene products were reported to be relevant for inflammatory responses by controlling infected cell death as a consequence of ROI generation.200,201 These mechanisms are separated from the direct antimicrobial effects of toxic radicals and will be discussed in the following. Intracellular pathogens may generate scavenger molecules to dispose of toxic radicals. Low molecular mass thiols, such as mycothiols from M. tuberculosis197 or homocysteine in Salmonella,202 are examples of ROI/RNI scavengers. In addition, both oxidative and nitrosative stresses are reflected in transcriptional changes in bacteria and initiation of DNA repair processes. Proteasome involvement in RNI resistance was reported for M. tuberculosis.203 Further, certain macrophage receptors interfere with NADPH oxidase function. Binding to CR1/CR3 does not induce respiratory burst and ROI production.204 The CRs, therefore, provide a relatively safe way of entry for intracellular bacteria.
Only gold members can continue reading. Log In or Register to continue