Aging and Atherosclerosis: Introduction
Atherosclerosis confers an illness burden on the general population larger than that associated with any other disease process. With advancing age, the incidence and prevalence of coronary, cerebral, and peripheral arterial disease (PAD) and aneurysmal formation are higher and their severity is more pronounced. Thus, cardiovascular disease (CVD) outcomes are worse in older compared to younger adults. Although age is the most powerful independent risk factor for CVD, the mechanisms by which aging predisposes to atherogenesis are only now becoming elucidated, as are specific strategies to optimally prevent and treat atherosclerotic disease in older adults.
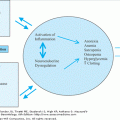
Research to date, spanning from basic science to epidemiology, suggests that aging potentiates atherosclerosis through a combination of factors (Figure 75-1). First, specific biological changes associated with aging increase vulnerability to atherosclerosis. Second, increasing age lengthens the time of exposure to known CV risk factors and modifies their effects. Finally, older individuals have increased comorbidities, which may contribute to or exacerbate the severity of atherosclerosis, particularly conditions that create or maintain a proinflammatory biologic milieu.

The clinical manifestations of atherosclerotic disease are more varied and severe in older compared to younger adults. Multiple etiologic factors have variable effects on the aging vasculature over a lifetime (Figure 75-2). Thus, vascular aging plays a primary and prominent role in predisposing older individuals to atherogenesis. This chapter highlights how vascular aging, in addition to both traditional and nontraditional risk factors, can enhance and accelerate the development of atherosclerosis in older adults.

Vascular Aging
It is a common misconception that vascular aging is synonymous with the development of atherosclerosis. Although the majority of individuals older than 60 years have clinically significant (>75%) coronary stenoses on autopsy, at least 25% of this age group will have only mild or no significant coronary plaque burden. Yet, age-associated vascular changes, and the cellular and molecular factors that underlie their development, do increase the vulnerability of older arteries to atherogenesis (Table 75-1).
LAYER | MOLECULAR | CELLULAR | STRUCTURAL | DYNAMIC |
|---|---|---|---|---|
Endothelium |
|
|
|
|
Intima |
|
|
|
|
Media |
|
|
|
|
The basic structure of the arterial blood vessel consists of three layers, each separated by a distinct elastic membrane. The intima is the innermost layer and consists of a monolayer of endothelial cells resting on a basement membrane made up of collagen and other extracellular matrix (ECM) molecules. The media consists primarily of smooth muscle cells (SMCs) on the background of an elastin-rich skeleton of ECM. Collagen, elastin, glycoproteins, and proteoglycans constitute the ECM. The outermost layer, the adventitia, contains a loose array of collagen fibrils as well as the vasa vasorum (blood vessels supplying the vasculature itself), nerve endings, fibroblasts, and mast cells.
Arterial structure changes with increasing age, but not in ways that necessitate atherogenesis. Morphologic changes associated with aging can include increased vessel diameter, intima-media thickening, SMC and collagen hypertrophy, fibrosis, and arterial wall calcification. In the absence of other predisposing factors, these progressive changes are age dependent but occur at varying rates in different individuals. Similar arterial alterations are seen prematurely in transplanted hearts and in a variety of vasculidities.
True atherosclerosis, by definition, requires the presence of an atheroma covered by a thin fibrous cap. An atheroma is a focal collection in the intima of foam cells (macrophages that have engulfed lipoproteins), proliferative SMCs (which have migrated from the media), and ECM. Postmortem studies of young Korean War soldier causalities revealed that the initial stages of atheroma formation can occur early in life.
Three major intertwining themes highlight how the biology of vascular aging predisposes to atherosclerosis (see Figure 75-1). First, exposure to various physical and metabolic stresses over time can alter normal pathways and processes. Second, progressive accumulation of metabolic byproducts can adversely affect arterial structure and function. Finally, time-dependent attrition of the basic arterial constituents and adaptive and reparative mechanisms of the arteries can lead to their dysfunction and, in turn, promote atherogenesis. This section details each of these themes.
Although biologic and chronologic ages are not necessarily equivalent, especially pertaining to vascular biology, the vector of time is inherent to the aging process (Figure 75-2). As such, chronic exposure to specific stresses over time contributes significantly to age-related pathology. These stresses include repeated exposure to both pulsatile and sheer forces on a mechanical level and ongoing oxidative stress at the molecular level. Stress-induced vascular changes ensue and, in some individuals, accelerate atherosclerosis.
Sheer stress over time provokes vascular injury, particularly at vessel bifurcations and curves, where atheromas tend to form. Endothelial cells subjected to chronic sheer stress can physically deform and display altered intracellular activity. Specifically, laminar sheer stress upregulates endothelial nitric oxide synthase (eNOS), the enzyme that produces nitric oxide (NO). Although sheer stress increases with age, overall eNOS activity is lower in aged compared to younger rats and contributes to impaired endothelium-dependent vasorelaxation. Cytokine-stimulated inducible-NOS activity, which raises NO levels, may create more negative feedback effect on eNOS than on inducible NOS. Age-related endothelial dysfunction is characterized by the decreased ability of smaller arteries to dilate in response to increased blood flow. Endothelial dysfunction in healthy older adults improves with exogenous nitrate administration, suggesting impairment, in part, of endogenous NO production. Endothelial cells play a key role in nearly every step of the atherosclerosis process, including plaque formation, platelet adhesion and aggregation, cellular migration, and proliferation. Thus, their dysfunction plays a key role in atherosclerosis. In a vicious cycle, atherosclerosis further impairs endothelial cell function (Figure 75-3).

Similar to shear stress, chronic biomechanical stress can also affect endothelial function by activating mechanoreceptors, thereby converting physical stimuli to biochemical signals. In vitro, endothelial cells exposed to repeated pulsatile stress have diminished eNOS expression and Akt and NO signaling. Pulsatile stretch also stimulates vascular SMC hypertrophy and proliferation. Such exposed SMCs synthesize ECM components in a disorganized fashion. Mechanical stretch also activates matrix metalloproteinases (MMPs), some of which are proliferative while others are destructive. Imbalanced activation of MMP subtypes creates adverse vascular remodeling, including fibrosis, which increases arterial stiffness and decreases elasticity, further exacerbating sheer and pulsatile stresses (Figure 75-3).
At the molecular level, augmented oxidative stress, measured by reactive oxidant species (ROS), is a constituent of both vascular aging and atherogenesis. Since its conception in 1950s, the free radical theory of aging has contended that metabolic production of oxidants, especially ROS, results in a wide array of adverse reactions involving nucleic acids, proteins, and lipids. Cumulative exposure to toxic oxidative effects leads to progressive cellular and tissue dysfunction and eventual disease and death. ROS are implicated in every step of atherogenesis, and this relationship is magnified in older vessels owing to ROS augmentation (Figure 75-4). Lipid oxidation is also accelerated by ROS and, in turn, stimulates many atherogenic steps including inflammatory cell activation, SMC, monocyte and macrophage growth factor release, reduction of eNOS-regulated platelet activity, metalloproteinase formation, cytokine release, and renin–angiotensin activation. Accordingly, oxidized low-density lipoprotein (LDL) facilitates vulnerable plaque rupture. Conversely, oxidized LDL receptor blockade inhibits atherogenesis.

With aging, the time vector permits accumulation of metabolic byproducts in the vascular wall, which creates adverse structural and metabolic sequelae. The prototypical example is accumulation of oxidized lipids in the bloodstream and their subsequent incorporation into atheromas. With time, the arterial wall also accrues increased amounts of normal and dysfunctional collagen, as well as broken and frayed elastin fibrils caused by elastase enzyme upregulation. Additionally, recurrent inflammatory processes result in vascular fibrosis. These alterations decrease arterial compliance to pulsatile flow, thicken the intima, and increase vascular permeability. Disruption in membrane permeability facilitates SMC proliferation and migration from the media to the intima as well as transit of immune and inflammatory cells that stimulate atherogenesis.
Progressive accumulation of advanced glycation end products (AGEs) also contribute significantly to CV structural and functional pathology over time. AGEs result from irreversible nonenzymatically formed bonds between glycated proteins. Time and high glucose concentration drive this reaction forward such in aging and diabetes. Long-lived proteins (collagen) are most susceptible to glycation over time and, once cross-linked, are less susceptible to routine hydrolysis and thus accumulate.
Structurally, AGE cross-linked arterial collagen is less compliant and contributes to age-associated central vascular stiffness. Similarly, AGE cross-linked myocardial collagen fibrils in the heart result in ventricular stiffness. In addition to their vascular structural impact, AGEs quench NO, stimulate ROS formation, facilitate inflammatory molecule and cell recruitment, and promote fibrosis. These alterations predispose the arterial wall to atherosclerosis. Receptors for AGE (RAGE) are located on endothelial cells, SMC, macrophages, and blood cells. AGE–RAGE attachment modulates a cascade of molecular events, resulting in vascular inflammation, fibrosis, vascular permeability, and altered NO signaling. Thus, time- and age-dependent accrual of AGEs, especially in diabetics, likely contributes significantly to age-associated increased endothelial dysfunction, vascular wall thickening, arterial stiffening, inflammation and fibrosis, restenosis, and atherogenesis. Clinical manifestations include isolated systolic hypertension (ISH), elevated pulse pressure (PP), left ventricular hypertrophy, delayed early diastolic relaxation, and atherosclerotic disease. LDL apoproteins are also a target of AGE, resulting in decreased LDL receptor uptake, more LDL oxidation, and increased susceptibility to macrophage engulfment, all leading to the formation of foam cells and atherosclerotic plaque.
The third age-related component predisposing to atherosclerosis is the reduced ability of reparative or compensatory biologic processes to appropriately adapt to the age-associated structural, dynamic, and molecular alterations. One hallmark of aging is cellular aging, also known as cellular senescence. Although the precise definition of cellular senescence is debated, it generally describes the irreversible arrest of the replicative cycle of a cell such that it no longer divides. The number of cell divisions in an organism is related to the organism’s expected longevity. In addition to exhibiting a flattened and enlarged morphology, senescent cells express a specific set of genes, including negative regulators of the cell cycle such as p53 and p16. Cells with shorter life spans have been cultured from individuals with premature aging syndromes such as Werner syndrome and Hutchinson–Gilford progeria syndrome, both of which are characterized by very early-onset atherosclerosis. Recent studies suggest that cellular aging also plays an important role in the development of age-related diseases in general.
Senescent forms of endothelial progenitor cells and vascular endothelial cells, which have been found in atherosclerotic plaques, are likely facilitators of atherogenesis. Factors that predispose these vascular cells to premature aging are numerous and involve the attrition of basic cellular constituents and molecular mechanisms. These factors include telomere dysfunction, faulty DNA repair systems, dysregulation of the insulin/Akt pathway, and altered angiotensin II signaling (Figure 75-5). Telomere dysfunction is of particular interest, since telomeres act as protective caps at the ends of chromosomes and are involved in cellular replication pathways. Shorter telomere length, a marker of cell turnover, has been associated with endothelial dysfunction, elevated PP, and the presence of traditional CV risk factors as well as clinically significant atherosclerotic disease. There are now data from at least one large clinical trial (WOSCOPS) showing that shorter telomere length is a predictor of coronary heart disease, independent from the effect of chronologic age.

Whether owing to telomere shortening and/or other factors, aging of vascular cells is associated with processes that lead to altered vascular structure and function and, in turn, accelerated atherosclerosis. Senescent vascular endothelial cells are found to have upregulated adhesion molecules, proinflammatory cytokines, and decreased NO production. Senescence of endothelial progenitor cells is associated with decreased angiogenesis and impairment of the complex vascular repair system intended to attenuate the chronic vascular injury and inflammation that leads to atherosclerosis. Targeting the mechanisms underlying vascular cellular senescence could represent new approaches to treating atherosclerosis in the future.
Vascular aging is characterized by specific structural and functional alterations that do not necessitate, but often potentiate, atherosclerosis (Table 75-1). Vascular aging changes result from ongoing exposure to physical and metabolic stresses, accumulation of metabolic byproducts, and attrition of basic cellular constituents and mechanisms that manifest as cellular senescence. Excess function or dysfunction of similar molecular pathways and cell types underlie vascular aging and atherosclerosis: endothelial cells, NO, ROS, SMC, AGE, and elements of the glucose metabolism pathway. Further elucidation of mechanisms that propagate vascular aging and its vulnerability to atherogenesis will facilitate development of interventions that could curtail both processes.
Risk Factors for Vascular Pathology in Older Adults
The age-related arterial structural and functional alterations that predispose older vessels to atherogenesis are heterogeneous across individuals and populations. This variability is influenced largely by the presence or absence of CV risk factors and comorbidities. Prolonged exposure to traditional CV risk factors over time impacts atherogenesis in older adults; diagnosing and modifying these risk factors early in life can reduce and prevent CV events in individuals up to 85 years of age. Since factors such as comorbidities, survivor bias, birth cohort effect, and long-term risk modification affect the potency of traditional and nontraditional risk factors in much older age, data regarding risk reduction in the very old are limited.
Age-associated increased central arterial stiffness results in hypertension being the most prevalent traditional risk factor in older adults: the lifetime risk of a 50-year-old developing hypertension is 90%. More than 80% of women and 69% of men older than 75 years are classified as hypertensive, and yet they have the lowest rates of control. Unlike in younger patients, who predominately have elevated diastolic blood pressures (DPBs), more than 60% of patients with hypertensions older than 65 years have ISH as defined by a systolic blood pressure (SBP) ≥140 mm Hg and a DBP <90 mm Hg. As individuals age, the CV risk index shifts from diastolic to systolic to pulse pressure (PP = SBP – DBP). This underscores the age-related difference in etiology (ISH is a manifestation of the vascular aging changes described in the previous section).
Although sphygmomanometry of the brachial artery is the most traditional noninvasive method to assess blood pressure, this technique does not fully characterize the age-related changes in central arterial stiffness. Brachial blood pressure measured this way represents an amalgamation of both small- and large-vessel properties. Accordingly, several noninvasive surrogate measures have been developed to assess central arterial pressure, including pulse wave velocity, carotid augmentation index, and estimated central pressures calculated by a generalized transfer function from applanation tonometry of a peripheral artery. These central artery stiffness surrogates are associated with increased CV risk, including myocardial infarction, stroke, renal disease, heart failure, and mortality. Ambulatory and nocturnal blood pressure results showing wide blood pressure variation also pose risk for adverse CV outcomes.
The importance of noninvasively determined central aortic pressure and its reduction was highlighted in The Conduit Artery Functional Endpoint (CAFÉ) study, which demonstrated discrepancies between two antihypertensive agents (amlodipine and atenolol) on CV outcomes despite similar brachial arterial cuff pressure changes. Improved CV outcomes associated with amlodipine compared with atenolol were coupled to an amlodipine-related reduction in estimated aortic stiffness.
Hypertension, itself, promotes vulnerability to atherosclerosis. This is especially true of the increased pulsatility associated with ISH. In response to increased perpendicular and parallel pressures on the arterials wall, vascular SMC hypertrophy and collagen accumulate. As discussed above, increased pulsatility on an endothelial cell with reduced capacity to stretch, such as in a stiff vessel, results in diminished NO signaling. Thus, hypertension exacerbates endothelial dysfunction and predisposes the endothelium to atherogenesis. As vascular collagen accrues in response to increased wall pressure so does AGE cross-linking, which, in turn, further thwarts vascular compliance in a viscous cycle. In this manner, hypertension and the excess forces it delivers to the older vascular wall accelerate vascular inflammation, AGE cross-linking, SMC hypertrophy, inflammatory cell migration, fibrosis, ROS production, apoptosis, and MMP stimulation, all of which predispose to atherogenesis.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


