57 Wilms Tumor
Wilms tumor is the most common intra-abdominal tumor of childhood and the second most common extracranial solid tumor among children. It is a malignant neoplasm of the kidney believed to arise from persistent embryonal remnants known as nephrogenic rests (Fig. 57-1).1,2 Management of Wilms tumor in the United States is based on the National Wilms Tumor Study (NWTS) group trials; similarly, in Europe the International Society of Pediatric Oncology trials have formed the basis of care for children with the disease. Wilms represents a paradigm of multimodality treatment and multi-institutional study. The joint efforts of pediatric oncologists, radiation oncologists, surgeons, and pathologists have resulted in a dramatic improvement in survival of children with Wilms tumor. Whereas the disease was once uniformly fatal, high cure rates have been achieved across all stages for patients with favorable histologic (FH) disease, the most prevalent subtype. Advanced stages of Wilms tumor with unfavorable histologic (UH) findings continue to pose major therapeutic challenges.
Epidemiology
Wilms tumor accounts for 6% of all childhood cancers in the United States, where the estimated incidence is 470 cases per year,3 or 8 per million children younger than the age of 16 years.2,4 In the United States, the disease is slightly more prevalent among girls than boys (1 : .92 for unilateral tumors, 1 : .6 for bilateral tumors). This disparity is not maintained worldwide; the global incidence is 1 in 10,000 children with no gender predilection noted.4 The incidence of Wilms is three times higher among Africans and African Americans than among East Asians, with the incidence among the European and American white population falling between these two extremes.5
Age at presentation depends on gender and laterality. Males tend to present earlier than females, and bilateral tumors earlier than unilateral tumors. The median age at diagnosis of bilateral disease is 30 months, whereas for unilateral disease it is 40 months.6 Seventy-five percent of all patients present before 5 years of age.7 Bilateral disease is noted in 4% to 8% of patients at presentation8–10; 7% to 12% of patients with unilateral Wilms demonstrate multifocal disease in the affected kidney.11
The majority of cases are sporadic, but Wilms tumor is thought to have a hereditary basis in 15% to 20% of affected patients.12 Approximately 1.5% of patients have familial Wilms, wherein one or more family members has the disease (usually siblings or cousins, rarely parents); in these cases, the mode of inheritance is generally thought to be autosomal dominant with variable penetrance.13
Etiology, Genetics, and Cytogenetic Abnormalities
Investigation of the role of parental environmental exposures as a causal factor of Wilms tumor has yielded inconsistent findings. Case-control studies have suggested that paternal exposure to lead and hydrocarbons increases the incidence of Wilms tumor in offspring, and that fathers employed as auto mechanics, welders, and machinists demonstrate an increased odds ratio for Wilms tumor among their offspring.14–17 However, other studies have failed to find any such correlation with paternal exposure or occupation.18–20 Previously reported maternal associations (smoking during pregnancy, hypertension of pregnancy, tea consumption) were not confirmed in a large case-control study.16,21
Genetic explanations of the causal factors of Wilms tumor originated with Knudson’s “two-hit hypothesis,” originally developed for retinoblastoma.22,23 Briefly, the two-hit hypothesis proposes that tumors develop as a consequence of two mutational events in a single cell. If the first mutational event is prezygotic (or germ line), the tumor is potentially hereditary and affected individuals are at risk of developing multiple tumors. Conversely, sporadic cases occur as a result of two postzygotic (or somatic) mutations in a single cell; because two postzygotic events are unlikely to occur in more than one cell, these patients are unlikely to present with multiple tumors.21 This hypothesis explains the earlier age of onset and bilateral presentation of familial Wilms compared with sporadic cases.13,24
Whereas the two-hit hypothesis has been confirmed in retinoblastoma to occur via inactivation of both alleles of a tumor-suppressor gene on 13q, the genetic events that lead to the development of Wilms tumor are less well-defined and appear to be more complex. Multiple genetic loci appear to play a role in the development of Wilms. The best known of these are located on the short arm of chromosome 11.25 Designated Wilms Tumor-1 (WT-1, at 11p13) and WT-2 (at 11p15) mutations in these putative tumor suppressor genes are found in 30% to 40% of Wilms tumors21 and are associated with three well-described congenital anomaly syndromes:
Although evidence for the role of these genetic loci in the development of Wilms is compelling, studies of affected pedigrees have failed to confirm either 11p13 or 11p15 as the location of the familial Wilms tumor gene.28 Based on epidemiologic and clinicopathologic analyses, NWTS group investigators have suggested that mechanisms other than germ line mutations (e.g., somatic mosaicism) are responsible for some bilateral and multicentric tumors.6,29
Other genetic mutations appear to contribute to adverse outcomes in children with Wilms tumor, although they are not clearly causative of the disease. Alterations in chromosomes 1p and 16q are associated with poor prognosis in children with Wilms tumor, implying that these genes are involved with tumor progression rather than tumor initiation21; hence they are referred to as Wilms tumor progressor genes, to distinguish them from WT-1 and WT-2. In a study of 232 children registered on NWTS-3 and NWTS-4, loss of heterozygosity (LOH) at either locus was associated with a poorer 2-year relapse-free and overall survival (OS), even with adjustment for stage and histologic subgroup.30
Heterozygous mutations in p53 are strongly associated with the anaplastic phenotype as compared with FH findings,31 suggesting that p53 abnormalities result in more aggressive histologic features. Overall, however, the incidence of p53 mutations in Wilms tumor is relatively low.32,33
Anatomy
Wilms tumors can arise in any area of the renal parenchyma. There is no predilection for either side. Extrarenal Wilms tumors are rare but can occur in the retroperitoneum, usually in the vicinity of the kidney. The tumor has occasionally been known to occur in the pelvis, inguinal region, or thorax, arising from displaced metanephric elements or teratomas.34
Pathologic Conditions
Childhood kidney tumors are organized according to the NWTS pathologic classification system (Table 57-1), which includes favorable as well as focal and diffuse anaplastic Wilms tumors, as well as clear cell sarcoma of the kidney (CCSK) and rhabdoid tumor of the kidney. Although these are histologically distinct from Wilms tumor, children with these diseases are included in NWTS trials because of the rarity of their occurrence, and treatment recommendations are based on NWTS findings.
Table 57-1 National Wilms’ Tumor Study Pathologic Classification System of Pediatric Renal Tumors
| Congenital mesoblastic nephroma |
Congenital mesoblastic nephroma, which is included in the NWTS classification system, falls at the benign end of the spectrum of pediatric renal tumors. It is the most common renal tumor of infancy,24 presenting at a median age of 2 to 3 months and occurring predominantly in males.35 It is characterized by bundles of spindle cells resembling fibroblasts or smooth muscle; cells at the periphery frequently extend deeply into the parenchyma or perirenal tissues. Despite its tendency for local extension, local recurrence or distant metastasis is highly uncommon, and the tumor may be curable by surgery alone (radical nephrectomy with wide margins). The cellular (or atypical) subclass of mesoblastic nephroma demonstrates increased cellularity and high mitotic rates. In older infants, this has been occasionally associated with local recurrence or the development of distant metastases.36 However, in infants younger than 3 months, cellular congenital mesoblastic nephroma is not associated with a poorer prognosis.37 Because there is no established role for radiation therapy in the management of mesoblastic nephroma, the subject is not discussed further in this chapter.
Wilms tumor is derived from primitive metanephric blastema and is classically triphasic in composition, consisting of varying proportions of blastemic, stromal, and epithelial elements.2 These elements appear to recapitulate normal renal embryogenesis. At the same time, Wilms tumors often contain tissues not found in the kidney, such as skeletal muscle, cartilage, and squamous epithelium, reflecting the pluripotent nature of primitive metanephric cells.21 Although triphasic composition is typical, Wilms tumor is characterized by tremendous structural and histologic diversity. Biphasic patterns are common, and tumors expressing predominantly or exclusively one element can still be categorized as Wilms tumor.38 Mixed composition is most common (41% of tumors), followed by blastemal predominant (39%) and epithelial predominant (18%); stromal predominant tumors are rare (1%). Although blastemal predominant tumors appear to be clinically more aggressive, these tumors are still classified as FH unless diffuse anaplasia is noted.39
Wilms tumors are classified as FH unless they meet clearly defined criteria for anaplasia. These criteria were established by Beckwith and Palmer40 following analysis of NWTS-1, and were refined by Faria and coworkers after analysis of NWTS-3 and 4 (Table 57-2). The original criteria did not account for location or multicentricity of anaplastic nests, and the distinction between focal and diffuse anaplasia lacked prognostic significance. In contrast, when Faria and colleagues’ strict criteria were applied in a review of NWTS-3 and four cases, patients with focal anaplasia were found to have a significantly superior 4-year survival as compared with patients with diffuse anaplasia; this difference was maintained stage for stage except for stage I tumors, in which survival was 100% for both focal and diffuse anaplasia.41 This prognostic significance highlights the importance of thorough sampling of the gross specimen during pathologic evaluation, as well as meticulous documentation of the exact specimen site from which each slide is obtained.
Table 57-2 Anaplastic Wilms’ Tumor:
| Definition of anaplasia | |
| Focal anaplasia: criteria | Anaplasia confined to a single region of the kidney |
| Diffuse anaplasia: criteria | Anaplasia present in more than one region of the kidney OR anaplasia in any extrarenal site (including vessels of the renal sinus, extracapsular infiltrates, or nodal or distant metastases) OR anaplasia in a random biopsy specimen |
Modified from the National Wilms’ Tumor Study-5: Therapeutic Trial and Biology Study. Unpublished. Activated June 16, 1995; last revised June 1998.
Eighty-nine percent of Wilms tumor cases in NWTS-3 were categorized as FH; 5% were anaplastic. The likelihood of anaplasia increased with age in NWTS-3; it was noted in only 2% of children younger than age 2 but increased to 13% in children ages 5 and older. Anaplastic tumors are significantly more frequent in black children than white children.42
Clear cell sarcomas and rhabdoid tumors of the kidney were originally believed to be monophasic variants of Wilms tumor; however, NWTS pathologists established both as unique tumors in 1978.40,43 They have since been analyzed separately but remain within the NWTS trials. Both entities are associated with strikingly poorer outcomes than Wilms tumor. CCSK is a primitive mesenchymal neoplasm distinguished by cells with poorly stained cytoplasm, prominent cytoplasmic vacuolation, and indistinct cell borders; groups of cells are often separated from one another by an arborizing network of thin-walled capillary vessels.24,39 CCSK accounts for 4% of all childhood renal tumors. Like Wilms, its most common site of distant spread is to the lungs; however, CCSK is distinguished by its propensity for bone and brain metastases as well. Twenty-three percent of children with CCSK in NWTS-3 developed bone metastases, as compared with 0.3% of all other children entered in the study.44,45
Rhabdoid tumor of the kidney, in contrast to CCSK, is marked by strongly acidophilic cytoplasmic staining. The tumor cells thus resemble myoblasts, but skeletal muscle markers are negative and the cell of origin is unknown.46,47 Like CCSK, rhabdoid tumors have a propensity for brain metastases; in addition, they can arise as primary CNS neoplasms,48 particularly primitive neuroectodermal tumors; this finding suggests a possible neural crest origin for rhabdoid tumors, a hypothesis that has been supported by histochemical analysis.47,49,50 Rhabdoid tumor occurs most commonly in male infants; median age at diagnosis is 13 months, and most cases are diagnosed in the first 2 years of life. In children older than 5 years diagnosed with rhabdoid tumor, pseudorhabdoid lesions (carcinomas, melanomas, histiocytic tumors, or myosarcomas) should be suspected.51 Rhabdoid tumors carry the poorest prognosis of all childhood renal tumors because of propensity for both local relapse and distant metastases.
Clinical Presentation
Eighty-three percent of children with Wilms tumor present with a painless abdominal mass.52 Other common presenting signs and symptoms include abdominal pain (37%), hypertension secondary to increased renin activity (25%), fever (23%), and hematuria (21%).53 Abdominal pain resulting from any cause (local distention, intralesional hemorrhage, peritoneal rupture) has been correlated with poorer 5-year survival.54 Acute subcapsular hemorrhage into the tumor results in a distinctive constellation of symptoms, including rapid abdominal enlargement, anemia, hypertension, and sometimes fever; plain radiographs of the abdomen may demonstrate a thin rim of eggshell-like calcification.55
Less commonly, children may present with varicocele, enlarged testicle, hernia, pleural effusion, congestive heart failure, or hydrocephalus.56 Several paraneoplastic syndromes have been reported in association with Wilms tumor, including Cushing syndrome, erythrocytosis, hypercalcemia, and acquired von Willebrand disease.57
Screening for children with known phenotypic or genotypic risk factors for the development of Wilms tumor is controversial. No randomized trials have been performed to address this question. Retrospective analyses of the NWTS registry and the National Childhood Cancer Registry of the United Kingdom failed to demonstrate a benefit to screening in children with aniridia, hemihypertrophy, or BWS.58,59 However, these studies were limited by the variation in incidence and time intervals of screening, as well as the methods used. In contrast, a case series analysis of children with BWS or hemihypertrophy, in which the case group had undergone systematic ultrasound screening at intervals of 4 months or less, suggested that screening sonograms every 4 months decreased the incidence of late-stage disease in these children (42% stage III to IV disease in unscreened children versus 0% in screened children, P < 0.003).60 A simulation model of sonogram screening three times yearly for children with BWS suggested that such a program would be comparably cost-effective to mammographic screening if applied from birth to age 4, largely because detection of earlier-stage disease would obviate the expense associated with radiation therapy.61 This hypothesis is supported by the finding that children with BWS in NWTS-3 and 4 were more likely to present with lower-stage tumors, although this may be a reflection of low biologic potential of Wilms tumor in these children in addition to the effect of increased surveillance screening.27
Routes of Spread
The most common location of local spread in Wilms is to the vessels of the renal sinus, which may be distended by tumor or demonstrate tumor thrombus. Penetration through the renal capsule is the second most common site of extrarenal spread. Not uncommonly, inflammation of the perirenal tissues can result in the formation of an inflammatory pseudocapsule, which may cause adhesions to adjacent organs such as the liver; yet on pathologic analysis, penetration of the true capsule by tumor is not demonstrated. Such cases are classified as stage I (limited to kidney, and completely resected, with negative margins).21 Even when tumor is adherent to or directly invades the liver, analysis of FH NWTS-3 cases demonstrated no detrimental effect on prognosis stage for stage.62 Conversely, patients with renal vein involvement were much more likely to have metastatic disease at diagnosis.63
Diagnostic and Staging Studies
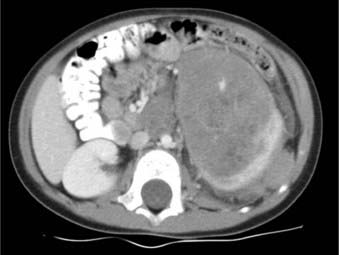
Abdominal ultrasound can fulfill several functions. It can establish the renal origin of the mass; demonstrate whether the mass is solid or cystic; and identify the presence and extent of tumor thrombus in the renal vein and IVC.34 Evaluation of the contralateral kidney is also possible, although this function is largely supplanted by CT. Contrast-enhanced CT imaging of the abdomen is recommended for further evaluation of the mass and adjacent viscera. CT can provide additional information on renal function, retroperitoneal lymphadenopathy, and extension to or invasion of adjacent organs, particularly hepatic metastases. Of note, however, in most cases in which CT identifies hepatic invasion, subsequent surgical exploration reveals only compression by tumor.64
The use of preoperative CT or MRI to evaluate for tumor in the contralateral kidney has been advocated in some surgical literature. However, others believe that imaging cannot replace intraoperative exploration.24 The NWTS protocols require surgical exploration to rule out contralateral disease. Debate also exists concerning the use of abdominal CT versus abdominal MRI in diagnosis of children with suspected Wilms tumor because of concern regarding radiation exposure.65
In the past, there has been controversy over whether lung metastases detected by CT scan but not chest radiograph warrant whole lung radiotherapy. This question was addressed by Meisel et al.66 in an analysis of NWTS-3 and 4 data. The authors found no significant differences in event-free survival (EFS) or OS among chest x-ray–positive, CT-negative children who received intensive doxorubicin-containing chemotherapy and whole-lung irradiation (WLI) versus multidrug chemotherapy (with or without doxorubicin) and no WLI. Although WLI resulted in fewer pulmonary relapses, a greater number of deaths were attributed to pulmonary toxicity. The authors concluded that WLI was not clearly necessary in treatment of children with discordant chest x-ray, CT findings,66 noting, however, that the small study numbers prevented a definitive recommendation.
Staging
Under the NWTS (now Children’s Oncology Group [COG]) system, the staging of Wilms tumor has continuously evolved to reflect prognostic information gained through successive clinical trials. The latest series of COG (AREN) protocols provide staging updated in 2008 (Table 57-3). The NWTS-3 and NWTS-4 staging system was significantly modified from NWTS-1 and NWTS-2, in which the categories were referred to as groups rather than stages. The NWTS-5 staging system was slightly modified from that used in NWTS-3 and 4 and the only change since NWTS-5 has been to upstage patients with spill or rupture confined to the flank, including tumor biopsy, to stage III.
Table 57-3 Wilms Tumor Staging