Keywords
Viral insertion sites, gene therapy, insertional mutagenesis, lentivial vectors, retroviral vectors, provirus, P140K MGMT
Introduction: Why we Should Know Viral Insertion Sites
Cancer gene therapy is an emerging field using genetic material to modify cells in vitro or in vivo to help effect a cure. In gene therapy, a therapeutic human-designed new gene (transgene) was often used to introduce to a subpopulation of human cells, such as human stem cells. For a cancer cell, the transgene can assist cell death or restore normal cellular functions, whereas for normal cells the transgene can protect them from drug-induced side effects and/or cytotoxicity. For instance, many drugs in cancer chemotherapy are DNA damage agents, specifically toxic to growing cells. Because cancer cells are fast growing, chemotherapy kills cancer cells very efficiently. However, many types of normal cells, such as bone marrow cells, also grow rapidly. Therefore, these therapies often cause severe bone marrow suppression, producing anemia, fatigue, easy bruising, and infections. Because of the side effects, doctors have to stop chemotherapy and the cancer therapy thus fails. To overcome these limitations, we recently began a gene therapy clinical trial at Case Western Reserve University in Cleveland, Ohio. The aim is to protect bone marrow of brain tumor patients who receive a high dose of chemotherapy. We expressed a human-modified DNA damage-resistant gene O 6 -methylguanine–DNA methyltransferase mutant, in which proline replaced 140 lysine (p140k MGMT ), in patient’s bone marrow . To achieve long-term effects, we used a lentiviral gene expression vector (from Lentigen, http://www.lentigen.com/ ) to transduce into patient’s hematopoietic stem cells. The genetic materials encoded by the lentivirus can be efficiently delivered into both dividing and nondividing cells . The p140k MGMT gene is semi-randomly integrated into the host human chromosome; thus, the inserted gene can be stably expressed in transduced hematopoietic stem cells. However, the viral insertion would cause mutagenesis. Every insertion of foreign DNA into a genome is in itself a mutagenic event. If a viral insertion is in a noncoding region and far away from any gene, the biological impact may be minimal. Nevertheless, if the insertion site is in or near a gene, the effect could be very serious. For instance, when the insertion site is in the middle of a coding sequence, it could disrupt the normal gene transcription, whereas if the insertion site is close to a gene promoter, it might alter the gene expression mechanisms. All of these conditions potentially lead to deleterious effects in patients. Indeed, in previous gene therapies using murine leukemia virus (MLV)-based retroviral vectors in X-linked severe combined immunodeficiency (SCID-X1) trials, five cases of lymphoid leukemia in a total of 30 patients (20%) were reported . A common finding in these patients was transcriptional activation of nearby proto-oncogenes such as the LIM domain only 2 ( LMO2 ) gene by the powerful enhancer elements contained within the retroviral long terminal repeats (LTRs) of the vector—a feature that has been difficult, in the case of SCID-X1, to recapitulate in animal models . For this reason, the U.S. Food and Drug Administration has since required all clinical trials using integrated viral vectors to examine and report the viral unique insertion sites (UIS).
The information on UIS is very significant. First, the location and distance of a UIS to a nearby gene indicate how it could affect gene function and expression. Second, if a UIS is detected with high frequency, it indicates a clone expansion, suggesting that the UIS might confer host cell growth advantage. Last, the viral insertion behavior could be modified. The viral insertion profiles would provide rich resources to develop better and safer viral vectors.
Viral Insertion Site Detection Methods
Polymerase Chain Reaction-Based Method: LAM-PCR
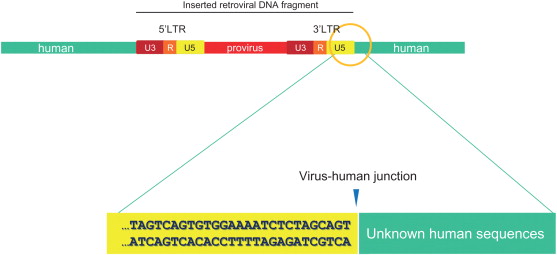
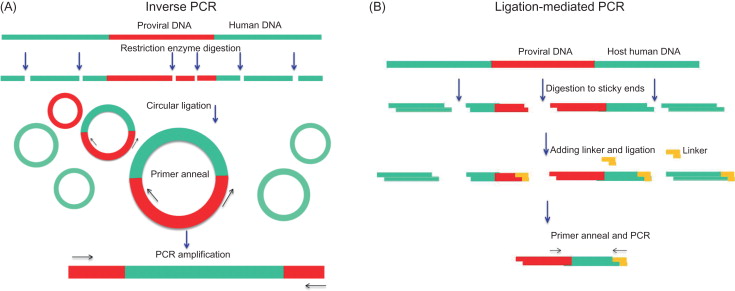
Once integrated into host cell genomic DNA, a viral DNA is called a provirus . Much effort has been made to develop methods to detect viral insertion sites. A viral insertion site or, more exactly, a viral vector integration site is composed of proviral sequences flanked by human genomic DNA ( Figure 3.1 ). For the virus–human junction, the proviral sequence is known, but the human sequences next to provirus are unknown, so a general approach is to use the proviral sequences as an anchor or starting point to walk into (identify) neighboring human sequences. After as little as 30–50 bp of the human sequences is found, it is rather easy to locate the position of the DNA in the human genome by the BLAST (Basic Local Alignment Search Tool) search engine ( http://blast.ncbi.nlm.nih.gov.easyaccess2.lib.cuhk.edu.hk/Blast.cgi ). Human genomes are gigantic, containing two of 3.3×10 9 bp DNA chains; by contrast, a proviral sequence is normally less than 5000 bp. Therefore, finding a UIS in human genomes is like finding a needle in a haystack. However, the retroviral integration has been well studied . The proviral DNA is flanked by LTRs. The LTRs are two 200- to 2000-bp repeated sequences, 5′ LTR and 3′ LTR ( Figure 3.1 ). Both of these contain a U3-R-U5 region, which is generated during virus reverse transcription . For lentivirus, the nucleotide sequence at 3′ LTR ends “… TGGAAAATCTCTAGCAGT” ( Figure 3.1 ). Thus, many methods have used the 3′ LTR end sequences to map the virus–human DNA junction. In 1981, Hayward first reported a method to isolate avian leukosis virus insertion site by a combination of Southern blot and genomic library screening . Several years later, much more powerful PCR-based methods were invented. Because a few hundred-base pair DNA sequences surrounding the virus–human junction would provide sufficient information, the fragment sizes are perfect for PCR reaction. In principle, the PCR method is able to efficiently enrich viral insertion site-containing DNA fragments by specifically amplifying sequences flanking the virus–human junction. Thus, it requires only a small amount of genomic DNA (~100 ng), and the detective sensitivity is very high. In general, there are two types of PCR-based methods. Inverse PCR ( Figure 3.2A ) uses restriction enzyme to first fragment genomic DNA and then ligate the DNA fragment into circular templates. The circles that contain the insertion sequence of interest are then preferentially amplified using primers, which point outward from the viral insertion (3′ LTR) sequence, usually 50–100 bp to the virus–human junction . Another method, ligation-mediated PCR ( Figure 3.2B ), uses a linker ligated to the ends of digested DNA. PCR is carried out with one primer against the insertion (3′ LTR) and another against the linker. Over time, ligase-mediated PCR has gradually become the most used method.
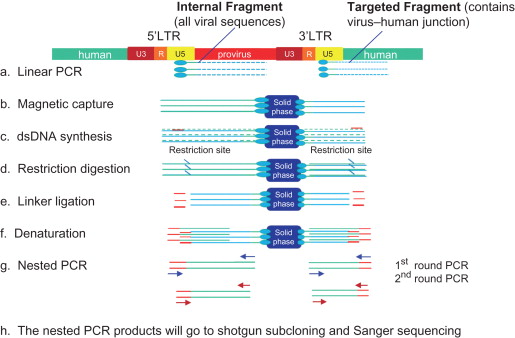
In 2002, Schmidt further modified ligase-mediated PCR by adding a linear PCR step and named it linear amplification-mediated PCR (LAM-PCR) . LAM-PCR has proven sufficient sensitivity, specificity, and robustness to enable the detection of viral insertion sites. LAM-PCR is illustrated in Figure 3.3 . Like other PCR methods, LAM-PCR focuses on the virus–human junction by designing a primer targeting viral 3′ LTR sequences. A unique feature here is that the primers are biotinylated, which allows them to tightly bind to magnetic beads. Nevertheless, linear PCR is not a typical PCR because the PCR reaction contains only one primer; therefore, the PCR products were single-strand DNAs. The biotinylated PCR products were subsequently isolated via the interaction of magnetic beads and a magnet device. This step is a very significant improvement because the population of viral insertion site-containing DNA fragments (usually approximately only 1 out of 10 7 DNAs) was physically separated from the rest of the genomic DNA. The single-strand PCR products were complemented to double-strand DNA by nonspecific hexanucleotide primer-mediated DNA synthesis. The newly formed double-strand DNA then digested with restriction enzyme and ligated to a linker. Thus, after adding the linker, each DNA fragment has virus 3′ LTR sequences at one end and the linker sequences at the other end, which are seamless for PCR. Then amplifications were done by two rounds of PCR (so-called nested PCR), which not only increased the production of the targeted DNAs but also increased the PCR specificity. The finalized PCR fragments consisting of viral vector–human genomic linker cassette sequences were shotgun cloned for Sanger sequencing.
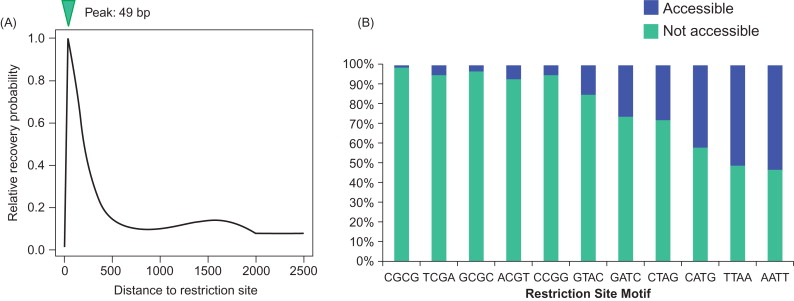
Although LAM-PCR has been used as the method of choice for viral insertion detection in gene therapy clinical trials for many years, the method suffers three major problems. First, the shotgun DNA cloning step is very labor-intensive and, more important, very inefficient. It can only pick up limited clones (<1000). The shotgun cloning itself is highly biased in selection of fragment size. For example, using Life Technologies’ TOPO TA cloning kit ( http://www.lifetechnologies.com ), small (<150 bp) PCR fragments had a much higher chance (>95%) than large fragments (>150 bp) of cloning into vectors . Even worse, the majority of small fragments actually do not have viral insertion sites. Most of them are primer dimers and nonspecific sequences. The second problem is the high degree of restriction enzyme bias. Due to the uneven distribution of restriction motifs, integration sites are most commonly recovered when they are approximately 49 bases from a restriction enzyme cleavage site, and frequency of recovery decreases sharply at longer or shorter distances ( Figure 3.4A ) . The fragments with integration sites far away from restriction cutting sites are thus less likely to be detected. Moreover, carefully designed studies have found that LAM-PCR works only for certain types of restriction site motifs ( Figure 3.4B ); for instance, LAM-PCR detection of a viral insertion site using a restriction enzyme motif containing “AATT” is 50% greater than that using “CGCG” . Therefore, selection of restriction enzyme has been very tricky and often multiple enzymes have been used. Third, the lineal PCR design is very inefficient. It is not a real PCR but, rather, a simple DNA extension, so 50 cycles of reactions can only generate a maximum of 50 copies of target DNA. For this reason, many more recently developed high-throughput methods dropped the lineal PCR step. Harkey used artificial mixtures of known retrovirally marked, single-cell-derived clones to systematically evaluate LAM-PCR detection ability and found that it failed to detect 30–40% of the clones, even with exhaustive analysis using multiple restriction enzymes .
High-Throughput DNA Sequencing
Clearly, LAM-PCR is still not good enough to meet the goals of viral insertion site detection in clinical trials. However, recently developed genomic technologies, especially next-generation DNA sequencing, have reshaped analytical methods for genetic studies. For example, the 454 massively parallel pyrosequencing technique was able to take a noncloned pool of PCR products and within 4 hr sequence approximately 1.3 million reads of approximately 800 bp in length each, producing a total of 560 million bp with a very low (0.04%) error rate . The DNA shotgun cloning step has been virtually replaced by the next-generation sequencing method in almost all late LAM-PCR studies . The result is truly remarkable. According to the Gene Therapy Safety Group Insertion Site report, in 39 of 40 studies that used old PCR methods without the pyrosequencing technique, the detected viral UIS was 49–739 per study. In contrast, 1 study using pyrosequencing showed more than 40,000 UIS. In a gene therapy clinical trial using the 454 sequencing method, Adair discovered 12,000 UIS in three tumor patients . Because 454 sequencing generates more than 1 million reads (sequences) at a reaction, the powerful 454 sequencing method not only largely increases the UIS detection speed and sensitivity but also is semiquantifiable. This feature gives clinical trials, which often require long-term follow-up (1–2 years), an additional edge to monitor the copy number changes of individual UIS. The data would provide vital information for the clone expansions or clone contractions .
Nevertheless, 454 sequencing is not yet perfect. The optimal DNA fragment size for 454 sequencing has been set at 400 bp. Sequencing longer or shorter fragments is less efficient. In fact, fragments less than 300 bp were removed even before sequencing. Paradoxically, the smaller fragment DNA in the step before nested PCR is more efficient. Small DNA lengths may account for a large fraction of the nested PCR products. Thus, the DNA size biases even in 454 sequencing are still not completely resolved.
Nonrestriction Enzyme Methods
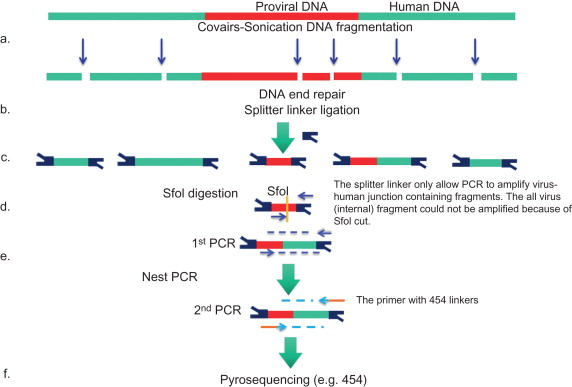
To eliminate restriction enzyme biases, at least three new restriction enzyme-free techniques have recently been developed. In 2011, Gillet reported an improved sonication DNA shearing (Covaris) method (see Figure 3.7 ) to fragment genomic DNA . The advantage of the DNA shearing method is that it generates entirely random and near equal-sized genomic fragmentations, thus avoiding the restriction enzyme bias problem. However, the sonication can damage DNA ends, causing difficulty for the linker ligation. Subsequently, an end-repair step is required. The second technique was introduced by LAM-PCR inventor Schmidt and is actually a continuing modification of his LAM-PCR protocols. To avoid restriction enzyme digestion, following the linear PCR step, the single-strand DNA is direct ligated to a single-strand linker with a special RNA ligase ( Figure 3.5A ). Thus, it was called nonrestrictive LAM-PCR (nrLAM-PCR) . This method has lower sensitivity than traditional LAM-PCR because of the low efficiency of single-strand DNA ligation . The third technique is transposase-mediated PCR. Brady used the bacterial transposase MuA to introduce a designed adaptor (Mu donor oligonucleotide) into human genomic DNA ( Figure 3.5B ). The adaptor functions as the linkers in LAM-PCR, allowing a PCR amplification between itself and the viral 3′ LTR . However, the sensitivity of this method is relative low—only 3382 UIS were detected in the study. This might be due to a low number of total transposase integration events. Moreover, the integration appears not to be completely random, more likely inserting at TTAA motif, although its overall integration bias was less than that of restriction enzyme. Thus, compared to LAM-PCR, the transposase MuA method did not show a notable advantage.
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Clinical Trials Using LV-P140K-MGMT for Gliomas
 Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
 Targeting Tumor Vasculature Using Adeno-Associated Virus Phage Vectors Coding Tumor Necrosis Factor-α
Targeting Tumor Vasculature Using Adeno-Associated Virus Phage Vectors Coding Tumor Necrosis Factor-α
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


