Herpesviruses
Herpes simplex type 1
Herpes simplex type 2
Cytomegalovirus
Varicella zoster virus
Epstein–Barr virus
Human herpesvirus 6
Intestinal viruses
Rotavirus
Norwalk virus
Adenoviruses
Astroviruses
Coxsackie
Caliciviruses
Respiratory viruses
Respiratory syncytial virus
Influenza
Parainfluenza
Metapneumovirus
Adenoviruses
Hepatitis viruses
Hepatitis A
Hepatitis B
Hepatitis C
Papovaviruses
JC
BK
Human papilloma
Other
Parvovirus
HTLV/HIV
West Nile
Herpesvirus Family
The herpesviruses are large double-stranded DNA enveloped viruses, which commonly infect humans. Typically, the primary infection is mild and self-limited and requires no therapy in the normal host. The human herpesviruses that cause infection are herpes simplex virus type 1 (HSV-1), herpes simplex virus type 2 (HSV-2), varicella zoster virus (VZV), cytomegalovirus (CMV), Epstein–Barr virus (EBV), and human herpesvirus type 6 (HHV-6). Human herpesvirus type 7 (HHV-7) has also been isolated, but its role in illness has not been elucidated. Human herpesvirus type 8 (HHV-8) gene sequences have been identified in tissue specimens of Kaposi’s sarcoma and some types of non-Hodgkin’s lymphoma, and an association with multiple myeloma has also been proposed.
Following resolution of the manifestations of the primary infection, lifelong latency can occur in normal hosts; the sites of latency are sensory nerve ganglia for HSV and VZV and leukocytes for CMV, EBV, and HHV-6. Endogenous virus can subsequently cause a reactivated infection in both normal and immunocompromised hosts. Generally in the normal host, reactivated infections have milder clinical symptomatology. All have been implicated as pathogens in patients with hematologic malignancies with the capacity to cause more severe clinical manifestations.
Herpes Simplex Virus
Patterns of Infection
Infections with HSV-1 frequently occur in early childhood. Transmission of virus occurs through contact with oral secretions. Transmission of HSV-2 occurs through contact with infected genital secretions, and thus infection generally occurs later in life, after adolescence. Clinical manifestations of HSV-1 include oral or nasolabial vesicles or ulcerations, keratoconjunctivitis, or encephalitis, whereas HSV-2 primarily causes genital vesicles or ulcerations. Serological conversion occurs with the primary infection, and the presence of IgG antibody provides an excellent indicator of prior infection and the presence of latent virus.
Immunocompromised patients are more susceptible for reactivation and potentially more severe manifestations [1] than normal hosts. In general, the intensity of chemotherapy regimens used in the treatment of hematological malignancy and the depth of depression of cell-mediated immunity correlate with both the risk for reactivation and the severity of the resulting clinical manifestations (Table 53.2). In seropositive bone marrow transplant (BMT) patients, the risk for recurrence is 70–80 % [2, 3]. In adult acute leukemia patients undergoing induction therapy, there is a similar 60–70 % frequency of reactivation [4]. In patients receiving ambulatory chemotherapy regimens for non-Hodgkin’s lymphoma, the risk is lower, in the range of 40–50 % [5, 6]. For comparison purposes, in patients receiving outpatient regimens for solid tumors (which are generally much less intensive than regimens used in the treatment of hematological malignancies), the recurrence rate is generally less than 10–25 % [7, 8].
Table 53.2
Rates of herpes simplex virus type 1 (HSV-1) and varicella zoster virus (VZV) infections in different patient groups
Group | Reactivation | |
|---|---|---|
Rates of HSV-1 infection (%) | Rates of VZV infection (%) | |
Bone marrow transplant | 70–80 | 20–50 |
Acute leukemia | 60–70 | 30 |
Hodgkin’s lymphoma | 40–50 | 15–20 |
Non-Hodgkin’s lymphoma | 40–50 | 5–10 |
Solid tumors | 10–25 | 5 |
Diagnosis, Prophylaxis, and Treatment
The manifestations of HSV-1 infection in patients with hematological malignancies are generally intraoral. Frequently, the labial lesions typically seen in the nonimmunocompromised host are absent and the oral ulcerations are indistinguishable from stomatitis due to tissue damage from cytoreductive therapy; thus, this distinction can pose a difficult diagnostic challenge. The concomitant occurrences of HSV infection and chemotherapy-induced mucosal damage result in a more severe form of mucositis [9]. The lesions tend to be larger, slower to heal, and more apt to become secondarily infected by bacterial opportunists. Atypical cutaneous satellite lesions may occur by autoinoculation of infected oral secretions into cutaneous abrasions. Extensive and deep oral ulcerations can lead to a greater susceptibility for bacteremia by α-streptococci, especially Streptococcus mitis, an organism that normally resides on the buccal mucosa [10]. Typically, HSV-2 infections cause vulval, intravaginal, or perianal ulcerations or vesicles.
Although the oral and genital mucosa are the primary sites of HSV infections, in the immunocompromised host extension to the esophageal and tracheal mucosa may occur [11–15].
Esophagitis can be caused by HSV alone or as part of a polymicrobial infection (especially Candida and CMV) [12, 16]. Endoscopic biopsy is important to distinguish various etiologies. Dissemination and involvement of visceral tissues can occur in profoundly immunocompromised patients [13–15, 17–19]. Herpesvirus pneumonia was noted to account for up to 5 % of nonbacterial pneumonias among allogeneic BMT patients [18, 20] in the preantiviral era.
Cultures of infected secretions or lesions permit confirmation of the diagnosis. Rapid methods using the shell vial technique coupled with antigen detection procedures [21] or using antigen detection immunofluorescent or immunoperoxidase assays alone [22–24] offer quicker and easier alternatives but may be less sensitive or specific. Cytologic examination of cells scraped from lesions using the Tzanck procedure can show the multinucleated giant cells caused by herpetic infection; this is rapid but relatively insensitive and cannot distinguish HSV from VZV. Detection of viral DNA by polymerase chain reaction (PCR) is an alternative to culture with similar sensitivity and specificity [25]. Although as noted serology can identify patients at risk for reactivation, it is not of value for the diagnosis of infection.
Typically the lesions from HSV infection occur 7–21 days after initiation of chemotherapy. This association with active therapy and its predictable temporal occurrence in seropositive patients has led to the development of prophylaxis strategies, as discussed next. In patients who are not being actively treated but who have severe deficiency of cell-mediated immunity due to progressive disease, malnutrition, cachexia, use of high-dose corticosteroids, or severe graft-versus-host disease (GvHD) after marrow transplantation and in patients with the acquired immune deficiency syndrome (AIDS), infection can occur at any time, and chronic progressive infections may occur.
Acyclovir, a purine analogue, is highly active against HSV types 1 and 2. Its phosphorylation by a viral-encoded thymidine kinase is necessary for its activation. Its poor phosphorylation by homologous cellular enzymes offers antiviral specificity. Further phosphorylation by cellular enzymes then occurs. Inhibition of the viral-encoded DNA polymerase provides additional antiviral specificity and accounts for its selective antiherpes activity. Finally, the triphosphorylated acyclic nucleoside is incorporated into the viral DNA chain, leading to chain termination and production of a defective virion.
Acyclovir has been demonstrated to be highly effective in both prophylaxis and treatment of HSV infections in patients with hematologic malignancies (Table 53.3). A variety of oral and intravenous regimens have been evaluated and found to be effective [3, 4, 26–32]. In treatment studies, shortening of the duration of viral shedding, time to pain relief, and interval until healing of lesions have been demonstrated. In prophylaxis studies, excellent control with few breakthrough infections has been noted. Because of its poor bioavailability (only 15 % after oral administration), caution should be exercised in patients who may have difficulty in tolerating oral medications, such as patients with severe mucositis or gastrointestinal toxicity, in order to ensure adequate plasma concentrations. Acyclovir is generally well tolerated but may be associated with nephrotoxicity if patients are not adequately hydrated or infusions are given too rapidly. Rare reports of reversible neurotoxicity, including tremulousness, lethargy, agitation, and disorientation have been reported; these toxicities resolve with discontinuation of the drug [33, 34].
Table 53.3
Treatment of herpes simplex virus infection with acyclovir
Viral and clinical response | ||||||
|---|---|---|---|---|---|---|
Parameter | Acyclovir [30] (IV) | Placebo (IV) | p a | Acyclovir [29] (PO) | Placebo (PO) | p a |
Median day (range) to | ||||||
Negative cultures | 3 (1–8) | 18 (4–49b) | 0.00005 | 2 | >9 | 0.0008 |
First decrease in pain | 6 (1–10) | 14 (1–14b) | 0.05 | 3 | 16 | 0.04 |
Cessation of pain | 10 (7–21) | 16 (7–49b) | 0.03 | 6 | 16 | 0.05 |
Crusting of all external lesions | 7 (2–28b) | 14 (6–49b) | 0.01 | 6 | 11 | 0.01 |
Total healing | 14 (6–28b) | 28 (10–49b) | 0.03 | 8 | 21 | 0.01 |
Valacyclovir, the l-valyl ester of acyclovir, is a prodrug that is metabolized to acyclovir within minutes of oral administration [35]. The bioavailability of acyclovir with this prodrug is approximately 50 % [36]. Plasma acyclovir levels are substantially higher and approximate those achieved with intravenous acyclovir [37]. Less frequent dosing is thus possible.
It is important to note that acyclovir is virustatic and suppresses viral replication only during the interval of drug administration. Following cessation of acyclovir, reactivation frequently occurs. When used in neutropenic leukemia and BMT patients, a course of therapy is generally given until neutrophil recovery. Recurrences after neutrophil recovery tend to be milder and self-limited and may not require retreatment. Often in nonneutropenic patients, treatment is given for 10–14 days. If severe immunodeficiency persists, lower dose maintenance therapy is sometimes advocated to prevent recurrence after the lesions are healed, to be continued until immunity is more robust.
There is considerable debate as to the relative merits of prophylaxis versus waiting until an established infection is documented before initiating treatment. Factors to be weighed in choosing between prophylaxis and treatment include the likelihood of infection, the depth of host immune deficiency, and the severity of infection if it occurs. Thus, while prophylaxis might be an acceptable strategy for BMT patients, in whom the risk for infections and morbidity are high, it would be of little use in patients with hematological malignancy not on active treatment, in whom the risk and morbidity of infection would be low. In BMT patients, it is currently advised to start acyclovir prophylaxis at the start of conditioning therapy and continue until engraftment occurs, resolution of mucositis, or approximately 30 days after HCT [28].
The issue of the emergence of acyclovir resistance is an important consideration in this regard. In BMT patients prophylaxis has been associated with a very low risk for drug resistance [38, 39]. In contrast, repeated treatment episodes of established HSV infection in BMT patients have been associated with progressively increasing rates of resistance [40]. An explanation for this apparent lower risk of resistance seen with prophylaxis has been suggested [41]. Certainly, the gradually improving host immunity that occurs with successful marrow transplantation and the relatively short duration of prophylaxis are contributory, because prolonged acyclovir prophylaxis in advanced AIDS patients, who have relentlessly deteriorating immunity, has been associated with a substantial risk for resistance. Initial reports of acyclovir-resistant HSV infection suggested that virulence had been attenuated and the clinical course was milder [42, 43]. However, reports of resistant isolates causing severe clinical illness have also appeared [44, 45].
For the most part acyclovir resistance has been mediated by alterations in the viral-encoded thymidine kinase production [42, 43, 46]. However, mutations in the DNA polymerase and altered substrate specificity for thymidine kinase are other potential modes of resistance [46]. Risk factors for the emergence of acyclovir-resistant HSV infections include severe GvHD and the lack of ganciclovir prophylaxis [47].
For patients with acyclovir-resistant HSV, treatment with foscarnet, a pyrophosphate analogue that directly inhibits viral DNA polymerase (and does not require thymidine kinase for its activity), may be effective [48]. In a randomized trial that compared foscarnet with vidarabine for acyclovir-resistant HSV infections in AIDS patients, foscarnet was both more effective and less toxic [49]. Another alternative is cidofovir. For patients with mild infection, another alternative approach would be to cease antiviral therapy to allow unassisted resolution. If progressive illness should ensue, then antiviral treatment could be initiated. Currently, there are no vaccines for either HSV-1 or HSV-2.
Cytomegalovirus
Cytomegalovirus infections are common in the general population, with serologic surveys demonstrating latent infection in 40–60 % of adults in most industrialized countries. Primary infection is often inapparent, but CMV can be a major cause of congenital malformations and hearing loss, mononucleosis, and sexually transmitted disease. Asymptomatic shedding in saliva and genital secretions is occasionally noted; clinical illness is uncommon unless the patient is immunocompromised.
Different strains of CMV have been identified by using two viral envelope glycoprotein genotypes (gB and gH). Some data suggest that infections by gB type 1 strains may be associated with greater survival than other gB types [50]. In another study, life-threatening myelosuppression was noted with gB3 and gB4 isolates, in contrast to gB1 and gB2 isolates [51]. Further studies are needed to ascertain whether characterization of strains may provide insights as to viral pathogenicity and prognostic information as to outcome from infection [52].
Patterns of Infection
Cytomegalovirus can be reactivated from endogenous latent virus or alternatively can be acquired from transfusion of blood products or from an organ graft. Leukocytes are a reservoir of latent virus; the risk for transmission can be reduced by depletion of leukocytes in blood products [53, 54] and avoidance of granulocyte transfusions [55]. Among seropositive and seronegative BMT recipients, the incidence of CMV infection was similar prior to the routine use of CMV seronegative blood product support [56]. Now infection rates are less than 10–15 % in seronegative recipients given seronegative blood products (although higher in the recipients of seropositive marrow grafts) [57].
In immunocompromised patients, CMV can be a cause of fever, leukopenia, thrombocytopenia, esophagitis, enterocolitis, interstitial pneumonitis, hepatitis, mononucleosis-like syndrome, chorioretinitis, and rarely, meningoencephalitis. Infection with CMV may predispose immunocompromised patients to sepsis [58, 59].
Although CMV excretion is a frequent occurrence in patients with acute leukemia undergoing intensive chemotherapy, its relationship to morbidity is poorly defined. However, several small surveys of acute leukemia patients suggest that the morbidity attributable to CMV may be underestimated [60–62]. Among leukemia patients, gastritis and esophagitis occasionally occur, and rarely, pneumonitis can be noted. In contrast, CMV can be the cause of life-threatening illness in BMT recipients.
The most common severe manifestations of CMV in BMT patients are interstitial pneumonitis (Fig. 53.1) and enterocolitis. Less serious manifestations are unexplained fever, esophagitis, gastritis, wasting, and hepatitis. Chorioretinitis is uncommon (in contrast to HIV-infected patients) [56]. Myelosuppression, manifest as delay in the recovery of counts after chemotherapy or unexplained cytopenias weeks to months following engraftment, can occur and can pose diagnostic challenges to distinguish from other causes of myelosuppression.
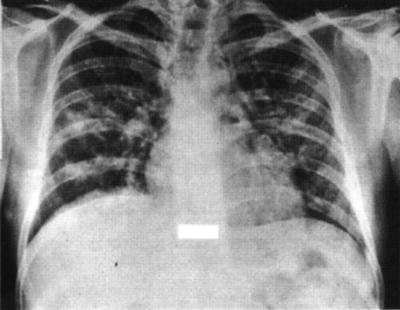
Fig. 53.1
Chest radiograph showing a diffuse mixed alveolar/interstitial pneumonitis due to cytomegalovirus
As with HSV, anti-CMV IgG antibody develops after primary infection. It provides a marker of endogenous latent virus. It does not appear that CMV-specific antibody protects against severe disease because several studies have not found a protective effect of high pretransplant antibody levels against infection [63]. Likewise, there is poor correlation between antibody responses and clinical responses [63]. Thus, while CMV antibody may have a contributory role in the control of CMV infection, it does not appear to be the most important response. Lymphocytopenia has been noted to be an unfavorable prognostic factor in BMT patients with CMV infection [64], and CMV-specific T-cell and natural killer (NK) responses have been noted following CMV infections [65–68]. The majority of survivors of CMV infection in BMT recipients develop either NK or T-cell cytotoxic responses or both, whereas few of those who succumb from infection develop cytotoxic responses and in the minority in whom cytotoxic responses could be found, the magnitude was much lower [65]. Thus, it appears that the early development of a robust cytotoxic response is crucial to resolution of CMV infection.
Although the development of cytotoxic responses is clearly immunoprotective, animal studies suggest that some cellular responses may be contributory to the pathogenesis of CMV pneumonitis [69–73]. Elucidation of which host immune responses are immunopathogenic and which are immunoprotective is crucial to the understanding of CMV pathogenesis and may permit insights into ways in which augmentation of certain responses and abrogation of others may be possible in future engineering of the constituents of the marrow graft through cell sorting techniques that discriminate various cell populations by their immunophenotypic differences.
Allogeneic BMT recipients are at greatest risk for serious illness from CMV infection, in contrast to autologous or syngeneic transplant recipients or nontransplant leukemia patients [74–79]. Among allogeneic recipients, risk factors for CMV morbidity include HLA disparity between donor and recipient, unrelated donor transplant, T-cell depletion, pretransplant CMV seropositivity, GvHD, older age, the use of immunosuppressive agents other than cyclosporine as GvH prophylaxis, viral excretion, and the intensity of the cytoreductive preparative regimens [20, 56, 57, 75, 79–85]. Detection of viral excretion from blood, urine, or throat secretions is indicative of active infection, and isolation of virus often precedes CMV disease by several days to 1–2 weeks. Viremia is most predictive of subsequent CMV disease [84]. Alternatively, recovery of virus from bronchoalveolar lavage specimens even in patients without any pulmonary symptomatology also identifies patients at high risk for the development of subsequent CMV pneumonitis [86, 87].
The CMV serostatus of the allogeneic BMT recipient is a significant factor in the long-term outcome. Positive CMV serostatus of the recipient remains a poor prognostic factor, especially in recipients of T-cell-depleted marrow or stem cells. An association of CMV with GVHD and nonviral infections or sepsis has been suggested as a possible mechanism [88]. A large study from the European Bone Marrow Transplant Registry demonstrated the importance of donor serostatus among CMV-seropositive recipients of unrelated grafts, possibly as a result of transferred CMV-specific immunity from the donor to the recipient [89]. In an analysis of a large cohort of T-cell–replete SCT recipients, both donor-positive/recipient-positive and donor-positive/recipient-negative recipients had a higher risk of mortality [90]. After controlling for neutropenia and CMV disease, only donor-positive/recipient-negative recipients had a higher risk of mortality. This was attributed to indirect immunomodulatory effects of CMV because there was an excess mortality due to bacterial and fungal infections when compared with donor-negative/recipient-negative recipients [90]. Collectively, these studies of the effect of CMV serostatus suggest that CMV infection before transplantation remains an important factor leading to poor outcome after transplantation, especially in recipients of T-cell-depleted transplants.
Although autologous BMT patients are at lower risk for CMV disease, such disease occasionally occurs. Patients receiving transplants for hematologic malignancy are at greater risk than those receiving transplants for solid tumors. Patients with multiple myeloma undergoing stem cell transplants in one study were at greater risk for CMV disease [91]. In one study, CD34 selection in autologous stem cell transplant recipients was associated with an increased risk for CMV disease [92]. However, this has not been observed by other groups [93].
Diagnosis, Prophylaxis, and Treatment
Cytomegalovirus has traditionally been detected by use of cultural methods [94]. Unfortunately, this is not very useful in treatment decisions because results may take several weeks. Modified cultural techniques using the shell vial method coupled with a rapid diagnostic antigen assay for the immediate early antigen permit demonstration of the virus within several days [95–97].
Newer assays include the demonstration of the pp65 antigen in leukocytes using immunofluorescence [98–100] or of viral DNA by the PCR [101–104]. Both the leukocyte antigen detection and PCR assays appear to permit detection of virus 7–10 days earlier than shell vial culture. Detection of virus DNA in plasma by PCR appears to be more sensitive than detection in buffy-coat leukocytes. Both the leukocyte antigen assay and PCR assays have the advantage of permitting quantification. Larger virus quantity may be more predictive of subsequent disease than less virus [105]. In one comparison, PCR appeared to be slightly more sensitive than the leukocyte antigen detection assay [106].
The gold standard for documenting CMV pneumonia in the past has been an open lung biopsy (Table 53.4). Because only half of the episodes of interstitial pneumonitis after marrow transplantation are due to CMV and the treatments are toxic, it is important to confirm the etiology at the outset. Similarly, for gastrointestinal syndromes attributable to CMV, tissue documentation is important to differentiate CMV from GvHD or other etiologies [107]. In contrast to patients with HIV infection, in whom CMV is frequently a pulmonary copathogen of less clinical importance, in BMT patients CMV typically can cause severe illness as a single pathogen. Transbronchial biopsy has not proved to be very reliable, yielding frequent false negatives. In contrast, bronchoalveolar lavage has been found to have a high sensitivity and specificity for documentation of CMV as an etiology of interstitial pneumonitis and is the usual method of diagnosis of CMV pneumonitis today [108]. This less invasive procedure has encouraged clinicians to be more aggressive in the evaluation of patients suspected of having pneumonitis and permits an earlier initiation of therapy.
Table 53.4
Interstitial pneumonia. Diagnostic procedures
Procedure | Yield (%) | Morbidity (%) |
|---|---|---|
Transtracheal aspirate | 5–10 | 2–5 |
Bronchoscopy | ||
Biopsy alone | 30–55 | 5–20 |
Bronchoalveolar lavage | 40–90a | 5–20 |
Transthoracic needle | ||
Aspirate | 17–60 | 10–30 |
Biopsy | 20–60 | 10–60 |
Open biopsy | 100 | 5–10b |
Untreated CMV pneumonitis after marrow transplantation has an 80–90 % mortality. A number of immune modulators and antiviral agents alone and in combination have been tested without success, including vidarabine, interferon, vidarabine plus interferon, acyclovir, acyclovir plus interferon, CMV immunoglobulin, ganciclovir, and ganciclovir with corticosteroids [109–115].
Ganciclovir, a nucleoside analogue similar in structure to acyclovir, differs from acyclovir in that it does not require phosphorylation by a viral-encoded thymidine kinase. Because human CMV does not encode for this enzyme, acyclovir has much less activity than ganciclovir against CMV. Phosphorylation of ganciclovir occurs by the phosphotransferase product of the viral UL97 gene. Although ganciclovir alone has had an antiviral effect in BMT patients, most patients derived no clinical benefit [114]. However, several nonrandomized trials demonstrated that the combination of ganciclovir with immunoglobulin results in clinical benefit, with survival rates of 50–70 % [116–119]. Although no controlled trials have been performed, this combination has been widely adopted for the treatment of CMV pneumonitis [120]. If therapy is delayed until ventilatory failure has ensued, outcomes are uniformly poor. Thus, early intervention is crucial. Following control of CMV pneumonia with ganciclovir and immune globulin, there can be occasional recurrences, and some clinicians advocate a maintenance course of ganciclovir for several weeks to several months following an episode of CMV pneumonia. Unfortunately, ganciclovir has not been shown to be similarly beneficial in the treatment of CMV-associated enteritis in marrow transplant patients [121]. A retrospective survey of the addition of intravenous immunoglobulin to antiviral chemotherapy did not appear to support the view that an additional benefit for an improved outcome is thereby obtained [122].
The onset of CMV pneumonitis in the marrow transplant recipient (prior to the antiviral era, discussed next) occurred approximately 2 months following transplantation. This was early after engraftment, frequently after the appearance of acute GvHD. Despite the gratifying improvements in the treatment of CMV pneumonia, many patients succumbed.
A number of investigators have sought ways to prevent either infection or illness from infection (Table 53.5). For CMV seronegative recipients whose donors are also seronegative, the use of CMV-screened blood products has been highly successful in minimizing the risks for CMV infection and disease [57, 98, 123–125]. Alternatively, the use of second-generation leukocyte filters for blood product transfusions similarly reduce the risk [54, 126].
Immune globulin, either CMV specific or from standard commercial lots, has also been shown to be beneficial in reducing CMV infection or disease [127–130]. It is not clear whether there is any advantage to using a hyperimmune globulin preparation compared to commercial lots of immune globulin not specifically selected for high titer anti-CMV antibody. Most of the efficacy from immune globulin has been demonstrated in patients who were seronegative prior to transplant. There does not appear to be any additive benefit of immune globulin to the use of filtered blood products in seronegative patients [122]. The magnitude of benefit from immune globulin has been less in seropositive patients [131], and immune globulin has been found to be of no benefit in autologous marrow transplant recipients [132].
Maribavir has been explored for the prevention of CMV infection in allogeneic stem cell transplant recipients. Despite encouraging results in pilot studies, a subsequent randomized trial showed no significant benefit with maribavir prophylaxis [133].
At present there are no satisfactory preventive measures for seronegative recipients who receive grafts from seropositive donors. Although immune globulin would be appealing in this situation, unfortunately it has not proved to be effective in reducing infection rates [123]. Prophylaxis with acyclovir or valcyclovir in combination with monitoring for viremia and treating viremia with preemptive therapy is advised by recently published consensus guidelines in this population [134].
In seropositive patients high-dose intravenous acyclovir given during the first month after transplantation has been associated with a reduced risk for CMV pneumonia [135]. In a three-arm trial, high-dose intravenous acyclovir during the first month, low-dose oral acyclovir during the first month, and high-dose intravenous acyclovir during the first month followed by 6 months of oral acyclovir were compared [136]. A reduced probability and delayed onset of CMV infection was noted in the group receiving high-dose intravenous acyclovir [136]. In this latter study, the prolonged course of oral acyclovir did not reduce CMV infection but did reduce viremia, and there was an improved survival benefit [136]. Similar acyclovir prophylaxis trials in solid organ transplant recipients show efficacy. Why acyclovir as prophylaxis seems to exert an anti-CMV effect whereas its use as treatment provides no effect is puzzling but may be indicative of a mild antiviral effect, which is demonstrable only when the viral burden is low as in the early stages of active infection and not apparent in later stages of infection, when the viral burden is substantially greater.
Ganciclovir has also been evaluated for prophylaxis (from engraftment to day 100) or as early treatment in BMT patients with active virus infection to preempt disease. As noted previously, asymptomatic patients with virus recovered from bronchoalveolar lavage fluid 35 days after transplantation, as well as those in whom viremia or excretion in urine or throat specimens occurs are at higher risk for CMV disease. Controlled trials have demonstrated a reduction in disease by initiation of ganciclovir at first detection of virus [86, 137]. An alternative strategy is to initiate ganciclovir at the time of engraftment in all seropositive patients [138, 139]. Because of its myelosuppressive toxicity, initiation of ganciclovir cannot be begun prior to engraftment. Even when it is begun shortly after engraftment, pancytopenia and interruptions of treatment are common, with an attendant risk of neutropenic infections. Patients with low marrow cellularity, elevated bilirubin, or increased creatinine are at greater risk [140].
The use of myeloid growth factors can lower the risk of myelosuppression. There is divided opinion at present as to the relative merits of prophylaxis versus early therapy [39, 141]. Several reviews have discussed the issues related to these strategies [39, 142–145]. It appears that prophylaxis is associated with fewer failures that could occur before or simultaneously with the first detection of active virus. However, the greater morbidity attendant on more frequent and more prolonged pancytopenia with ganciclovir started at the time of engraftment makes ganciclovir less well tolerated than when started later. Moreover, early therapy given only to patients with active infection spares a significant proportion of patients the toxicities and cost of antiviral therapy. The greater use of the leukocyte antigen and PCR assays to detect virus earlier than with the shell vial culture assay may reduce some of the failures associated with early therapy, making this more advantageous than prophylaxis [39]. If ganciclovir is used for prophylaxis or as early therapy, there appears to be no added benefit for acyclovir during the first month [146].
In an attempt to reduce the toxicity of long-term ganciclovir, several variations of daily therapy have been attempted. Ganciclovir three times per week was not found to be adequate to prevent CMV reactivation, at least in T-cell-depleted marrow transplant recipients [147]. On the other hand, a preliminary report suggests that 3–6 weeks of daily ganciclovir may be as efficacious as a longer course while causing less toxicity [148, 149].
Oral ganciclovir is useful in AIDS patients for maintenance therapy to prevent recurrence of CMV retinitis, but because of poor bioavailability, serum concentrations are lower and breakthrough infection rates appear to be higher [150]. Emergence of resistance has also been noted and appears to be greater than with intravenous ganciclovir [151, 152]. Valganciclovir, a prodrug of ganciclovir, is metabolized to ganciclovir and has excellent bioavailability [153–155]. Plasma concentrations are quite high. Valganciclovir has been shown to be an alternative to ganciclovir, which may make prolonged courses of therapy more feasible [156–158].
Resistance to ganciclovir occurs through mutations in the CMV UL97 gene [159–161]. DNA polymerase (UL54) mutations can also occur, resulting in high-level resistance [161]. Although many ganciclovir-resistant CMV mutants have mutations in the UL54 gene (DNA polymerase) as well as in the UL97 gene, susceptibility to foscarnet is retained [161–163]. Although rising CMV antigen titers can be seen in approximately 40 % of patients early on with ganciclovir maintenance therapy, most isolates are susceptible to ganciclovir, indicating that host factors and medications, rather than resistance, accounts for the rise in titers. Additionally, no correlation was seen between CMV titers and CMV disease [164].
Foscarnet, a pyrophosphate analogue, targets the viral DNA polymerase. It represents an alternative to ganciclovir both as treatment and as prophylaxis [165–169]. Sparing of the blood counts makes foscarnet an attractive alternative to ganciclovir in patients in whom marrow reserve is marginal. However, nephrotoxicity and renal wasting of electrolytes are prominent side effects of foscarnet. In one small study, the combination of foscarnet and ganciclovir was effective and may have less treatment toxicity [170]. Foscarnet is an option for ganciclovir-resistant CMV [171].
Cidofovir is a nucleoside analogue with potent anti-CMV activity not requiring phosphorylation by the UL97 gene product, which acts against the viral DNA polymerase (UL54). It is an effective treatment for CMV retinitis in HIV-infected patients [172–175]. Advantages include less frequent dose intervals (once weekly at first, then every other week). Limitations include considerable nephrotoxicity (which in some cases was irreversible) and iritis. Concomitant probenecid and intravenous hydration are necessary. There are to date only limited data on BMT patients [163]. Because many ganciclovir-resistant CMV mutants have alterations not only in the UL97 gene but also in the UL54 gene (DNA polymerase), resistance to cidofovir can also be present [161, 162]. Accordingly, foscarnet is the preferred antiviral in the face of ganciclovir resistance.
Historically, most episodes of CMV pneumonitis occurred during the first 100 days after marrow transplantation. However, with the advent of antiviral prophylaxis in recent years, late-onset CMV pneumonitis has increasingly been noted beyond 100 days [176–182]. In part this is due to the increasing use of unrelated and HLA-mismatched donors, where immune reconstitution is much slower than after allogeneic transplants from genotypically identical siblings. However, delay in the development of cytotoxic anti-CMV responses in patients in whom ganciclovir has been used has also been noted [183, 184]. Most late-onset cases occur in patients in whom CMV infection was documented during the first 100 days [185]. The risk of late-onset CMV pneumonitis appears to be greatest in patients with chronic GVHD. This is a group in whom CMV surveillance may be particularly important. An unpublished trial testing whether ganciclovir prophylaxis late after BMT in patients with risks for late CMV disease demonstrated it to be safe and reduced viremia but no substantial reduction in CMV disease or survival benefit.
With recognition of the importance of development of cytotoxic cellular responses to CMV for resolution of infection as well as of the ability to clone lymphocyte populations and expand them ex vivo with interleukin-2 and repetitive CMV antigenic stimulation, it has become possible to consider cellular immunotherapy of CMV infection [186–189]. Consideration of donor immunization may also be important because recipients of grafts from seropositive donors may have earlier recovery of cytotoxic responses than recipients of grafts from seronegative donors [183]. Such immunotherapeutic approaches may prove to be increasingly important in years to come as the emergence of ganciclovir resistance among CMV isolates increases [190].
Because the risk of CMV disease is substantially greater in seropositive than in seronegative patients, newly diagnosed leukemia patients in whom a transplant is contemplated after induction therapy should be given only CMV-seronegative blood products throughout their induction regimen if they are seronegative. The value of CMV-negative blood products in autologous transplantation patients, in whom the risk for serious CMV disease is low, is questionable but is recommended by some [191].
Immunization of the donor or recipient by a live attenuated, killed, or subunit recombinant CMV vaccine might be attractive in speeding restoration of immunity [192]. However, although an attenuated vaccine has been found to be immunogenic in solid organ transplant patients, concerns regarding its safety in more severely immunodeficient marrow transplant patients have prevented trials of a live vaccine. A glycoprotein or DNA vaccine would be desirable. Clearly, abrogation of the recipient’s immunity by the preparative regimen may mean that prior immunization of the donor as well as repeated boosting of the recipient is necessary to establish meaningful immune responses.
Varicella Zoster Virus
Patterns of Infection
Varicella zoster virus, like the other herpesviruses, can be a significant pathogen in patients with hematologic malignancies. Varicella (chickenpox) is the primary form of infection, and zoster (shingles) is the reactivation form of illness. The initial portal of entry is uncertain, but the respiratory tract, skin, and conjunctiva are candidate sites. After local replication, viremia occurs with dissemination to multiple cutaneous (and visceral) sites. The skin lesions begin as erythematous macules, progress to papules and then to vesicles (with clear fluid), which evolve to pustules (with cloudy fluid containing interferon and leukocytes), and flatten to scabs, until finally healing takes place. The hallmark of infection is the vesicle. Crops of vesicles classically exist on a common erythematous base. Scarring is unusual in varicella, but the inflammatory reaction in zoster may be more severe with greater pain and scarring. Zoster characteristically is dermatomal, and lesions typically stop at the midline. In severely immunocompromised patients extension to adjacent dermatomes may occur, the vesicles may become confluent, and cutaneous dissemination can occasionally occur. Approximately 5–10 % of patients with non-Hodgkin’s lymphoma, 15–25 % of patients with Hodgkin’s disease, and 20–50 % of patients undergoing BMT develop zoster [193–198] (Table 53.2).
In the normal host varicella and zoster are self-limited. In patients with hematologic malignancies, varicella may be quite severe and life threatening. In the preantiviral era, varicella frequently progressed to visceral dissemination and death [199].
Untreated dissemination occurs in 30–40 % of marrow transplant recipients [195, 196] and in up to 20–30 % of children treated for hematological malignancy [199]. Visceral dissemination is a severe manifestation and pneumonia can be particularly deadly. In marrow transplant patients an intra-abdominal presentation can occur, with severe abdominal pain [200–202]. This can be particularly challenging to diagnose because it may occur in the absence of cutaneous vesicles. A high index of suspicion should be maintained because if the condition is not treated promptly, it can result in a high mortality. Manifestations of pancreatitis, hepatitis, and peritonitis are frequent, and in patients in whom laparotomy was inadvertently performed, inflammatory changes, vesicles on serosal surfaces, and mesenteric adenitis have been noted.
The onset of VZV infection in the marrow transplant recipient is much later than that of the other herpesviruses, occurring an average of 5 months following transplantation [195–198] (Fig. 53.2). Susceptibility for reactivation can persist for many months, especially among patients with chronic GvHD [195, 196]. A study of humoral and lymphocytic proliferative responses to VZV in BMT recipients indicated that in approximately 26 % of patients a subclinical reactivation occurred [203]. Similarly, in patients undergoing induction therapy for leukemia, 5–10 % subclinical reactivation occurred. Thus, when both clinical and subclinical infections are considered, VZV infection is a common occurrence in BMT recipients.
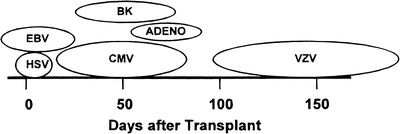
Fig. 53.2
Viral infections and their time course after marrow transplantation. HSV herpes simplex virus; EBV Epstein–Barr virus; CMV cytomegalovirus; BK BK virus; ADENO adenovirus; VZV varicella zoster virus
Among patients treated for lymphoma, VZV infection is more common in those with advanced disease, those treated with combined-modality chemoradiotherapy [193], and in patients who have received rituximab [204]. It appears that the frequency of zoster is increasing as more dose intensive therapies are being employed. Risk factors for VZV in allogeneic marrow transplant patients include acute or chronic GvHD, older age, and the posttransplant use of antithymocyte globulin. Among autologous transplant patients, the underlying disease of Hodgkin’s or non-Hodgkin’s lymphoma is a risk factor [197]. Zoster develops more commonly in dermatomes of prior radiotherapy.
Diagnosis and Treatment
Diagnosis can usually be made solely by visual inspection of the cutaneous lesions. Examination of the cellular contents of a vesicle can demonstrate multinucleated giant cells on Tzanck smear [25]. Culture can be confirmatory but special handling must be used for this heat-labile cell-associated virus. Immunofluorescent staining can also be used [205]. PCR techniques may offer yet another alternative [25].
Acyclovir is highly active against VZV [206–208]. Higher concentrations are required to inhibit VZV in vitro as compared to HSV-1 and HSV-2. Generally, plasma levels achieved with standard doses of acyclovir given orally are insufficient to achieve inhibitory plasma concentrations. High doses of oral acyclovir (800 mg given five times daily) achieve peak plasma concentrations that approximate in vitro inhibitory concentrations for most isolates of VZV and have been found to be useful in nonimmunocompromised patients in whom treatment is initiated early after onset of infection [209]. The adequacy of this dose in compromised patients has not been well demonstrated, although one small trial in BMT recipients suggested that a beneficial effect was achieved [210]. However, because only a small number of patients have been studied, intravenous acyclovir is preferable as initial therapy unless the infection is mild and the patient can be closely monitored to institute parenteral therapy if there is progression or no response.
Vidarabine in the past was an alternative treatment [211], although controlled trials have shown acyclovir to be superior [208]. Interferon-α has also been shown to be efficacious [212], although it has been evaluated only in localized disease and there is no published experience of its use in disseminated disease [213].
Several oral agents are effective therapies in nonimmunocompromised patients. These include famciclovir and valacyclovir, prodrugs of penciclovir and acyclovir, respectively [214–216]. These agents have greater bioavailability and longer half-lives than oral acyclovir, which make them excellent candidates for outpatient treatment. Their efficacy has not been shown to be suitable for zoster therapy, including in immunocompromised patients [217, 218]. They seem to be suitable for mild to moderately severe cases of zoster when the patient can be observed carefully or as a change from intravenous acyclovir once an initial response to therapy is achieved.
The risk for acyclovir resistance in VZV is much less than in HSV. However, several reports have noted the emergence of drug resistance [219, 220]. As with HSV, the mechanism of resistance appears to be an altered thymidine kinase, and thus foscarnet is a suitable alternative for resistant pathogens [220].
Immunization and Prophylaxis
A live attenuated vaccine has been evaluated in normal and immunocompromised children with leukemia and found to be beneficial in reducing the risks for VZV infection in patients with ALL in remission for more than 1 year [221–224]. Because of the risks of dissemination by the vaccine strain in patients with severe immunocompromise, its role in such patients is limited [225]. On the other hand, family members of seronegative children with leukemia could be immunized to reduce the risk of the patient contracting a primary infection [226].
Because of the high mortality of VZV associated disease among severely immunocompromised HCT recipients and the lack of clinical data, passive immunization with either VZIG or VariZIG is advised under very specific circumstances [134]. For VZV-seronegative immunocompromised HCT recipients, VZIG or VariZIG should be administered within 96 h after close or household contact with a person with chickenpox or shingles. VZV-seropositive patients who are highly immunocompromised can be given VZIG or VariZIG after exposure to VZV, including after exposure to a VZV vaccine and developing a varicella-type rash. Postexposure acyclovir or valacyclovir may be used as an alternative if VZIG or VariZIG is not available [227, 228].
Acyclovir given orally in intermediate doses for a number of months has been shown to reduce the risks for VZV infection following marrow transplantation [203, 229–233]. Administration of acyclovir for 1 year in BMT recipients has been shown to be effective without a rebound of zoster disease after discontinuation [234]. In one study, the possible transfer of VZV-specific T-cell immunity from donor to recipient in the marrow transplantation setting was evaluated [235].
Unfortunately, simple transfer of immunity was not evident. Whether boosting the donor immunity prior to adoptive transfer might improve on this remains to be seen. Currently, it is advised that autologous and allogeneic transplant recipients receive acyclovir prophylaxis for 1 year after transplantation [134]. In patients who have cGVHD or are on chronic immunosuppression, a longer duration of prophylaxis is required.
Epstein–Barr Virus
Pathogenesis and Immunity
Epstein–Barr virus is an ubiquitous organism with the majority of adults being infected. Transmission is usually via oral secretions. Sites of virus replication include the oropharyngeal epithelium and B lymphocytes, the latter being the major reservoir of endogenous latent virus [236–239]. Primary infection is often subclinical, but in adolescence EBV can cause the syndrome known as infectious mononucleosis. It can also cause acute or protracted fever, malaise, fatigue, autoimmune hemolytic anemia, cytopenias, rash, or adenopathy [239–241].
Rarely, it has been associated with several varied neurologic syndromes. It has been implicated as a factor in the pathogenesis of African Burkitt’s lymphoma, central nervous system lymphomas, polyclonal and monoclonal lymphoproliferative disorders in solid organ transplant patients, nasopharyngeal carcinoma, and lymphoproliferative disorders associated with the X-linked lymphoproliferative syndrome; in addition, a possible role in Hodgkin’s disease has been proposed [239, 242–245].
Primary infection elicits both humoral and cell-mediated immune responses [239, 246–249]. During the acute illness, the production of heterophile antibody and autoantibodies occur. The heterophile antibody (antibody to sheep erythrocytes not absorbed by guinea pig antigen) is most commonly used to document acute infection. Early after infection, IgM antibody and IgG antiviral capsid antibody (VCA) appear, along with antibodies to the early antigen (EA). IgM antibody disappears generally within 2–4 months, but anti-EA antibody may persist for several months or longer. Anti-EBNA antibody appears late, several months after the acute infection, and persists for life. Host cellular responses include natural killer cells and EBV-specific cytotoxic and suppressor T cells. The atypical lymphocytosis characteristically seen during infectious mononucleosis is primarily made up of the responding T-cells rather than the infected B-lymphocytes, and these represent the host attempt to control B-cell lymphoproliferation.
Within the B lymphocyte, the EBV genome persists as episomes or occasionally becomes integrated into the host cellular genome. In latently infected cells EBV gene expression is restricted to nuclear protein genes (EBNA), a membrane protein (LMP), and EBV-encoded RNAs (EBERs), which have roles in maintenance of latency, transformation, and prevention of apoptosis [239]. Although latent EBV infection confers a proliferative advantage to B cells, by itself it does not appear to be sufficient for oncogenesis. Rather, other events involving alterations in oncogenes such as c-myc or N-ras or mutations in tumor suppressor gene such as p53 or RB appear to be necessary for the transition from polyclonal proliferation of B lymphocytes to a monoclonal B-cell malignancy [250, 251].
Diagnosis and Treatment
Culture of EBV is problematic because of the lack of readily available assays in diagnostic virology laboratories. However, in research laboratories EBV can be identified by its ability to transform cord lymphocytes [252]. Today, assays for viral DNA detection are mostly straightforward and monitoring by quantitative PCR is readily available [253, 254].
Epstein–Barr virus can be detected in salivary secretions in 15–20 % of the general population [252]. Excretion rates are higher in immunocompromised patients. Patients receiving immunosuppressive therapy have excretion rates of about 35 %, patients with malignancies have excretion rates in the range of 40–50 %, renal transplant patients 45–60 %, leukemia patients 75–100 %, and HIV-infected patients 45–70 % [255–260]. In adults, most infections occur in previously seropositive patients, whereas in children both primary and reactivated infections occur. The number of EBV genomes detected in peripheral blood leukocytes is increased in immunocompromised patients.
Excretion rates vary considerably according to the intensity of immunosuppressive therapy and the extent of depression of host cell-mediated immunity. Thus, patients with repeated organ graft failure, those who receive multiple immunosuppressive drugs, and those who receive antithymocyte globulin are especially vulnerable for reactivation. Similarly, depending on the immunologic defect, the serologic responses to EBV infection may be more or less pronounced than in immunocompetent individuals. Frequently, serologic responses are exaggerated [261], whereas the more important cell-mediated immune responses are generally suppressed.
Although EBV shedding may be asymptomatic, several EBV-associated syndromes have been identified in patients with hematologic malignancies. Oral hairy leukoplakia commonly found in AIDS patients is also noted in patients with hematologic malignancies, especially those undergoing marrow transplantation [262–264]. There has been increasing recognition of EBV-associated lymphoproliferative syndromes in transplant recipients [255, 265–267]. Children appear to be at greater risk than adults, and seronegative patients are at substantially greater risk than seropositive patients. The risk also varies according to type of transplant, with heart-lung transplant recipients appearing to be at greater risk than those with renal, liver, or cardiac transplants. The evidence for a role of EBV as a causative agent includes the presence of EBV DNA and EBV-specific proteins within tumors, the presence of activation markers consistent with latent EBV infection (CD23 and adhesion molecules), the association with primary infection, and the analysis of terminal repeats indicating monoclonality. Several types of posttransplant lymphoproliferative disease (PTLD) have been described. A mononucleosis syndrome that is localized and self-limited is the least serious. A more widespread mononucleosis syndrome, which can be progressive, and lymphoma, which can be extranodal involving the gastrointestinal tract, brain, and other organs, can also occur. Several classification schemes have been proposed, including one which incorporated molecular genetic analyses with morphology [250].
In BMT recipients, EBV infection has been characterized both serologically and by EBV detection [268, 269]. Reactivation of EBV in seropositive recipients is frequent and generally occurs 2–4 weeks following transplantation. Clinical manifestations have not been associated with viral reactivation, however.
Infrequently, PTLD has been noted in marrow recipients, generally in the setting of severe GvHD, the use of antithymocyte globulin or anti-T-cell antibody, or after T cell-depleted, cord blood, or haploidentical donor transplantation [270–277]. In a comprehensive review of more than 18,000 patients who had undergone allogeneic BMT, PTLDs were noted with a cumulative incidence of approximately 1 % at 10 years [278]. Most cases occurred between 1 and 5 months posttransplant with few cases occurring after 1 year. In a multivariate analysis, lymphoproliferative disorders within 1 year of transplant were associated with unrelated or mismatched donor transplants, T-cell depletion of the donor marrow, the use of either antithymocyte globulin or antiCD3 monoclonal antibody as part of the GvHD prophylaxis regimen, acute GvHD, and conditioning regimens that included radiation. Methods of T-cell depletion methods that did not remove B cells (the target cell of EBV infection) were associated with a substantially higher risk than methods that depleted both B and T cells. The only risk factor for late-onset PTLD was the occurrence of chronic GvHD [278].
Although most EBV-associated lymphoproliferative disorders have been described in solid or BMT recipients or in HIV-associated immunodeficiency, the increasing use of potent purine analogues, such as fludarabine or cladribine in combination with corticosteroids, has created a new population of oncology patients with profound cell-mediated immunodeficiency. A recent study of EBV DNA load in leukemia and lymphoma patients receiving fludarabine, cyclosphosphamide, and dexamethasone indicated a substantial increase in viral load in some patients [279]. Although no EBV-associated lymphoproliferative disorders have been noted to date, these are cautionary observations, which warrant further study.
Acyclovir is active against EBV both in vitro and clinically [280–283]. Its clinical efficacy in nonimmunocompromised patients with infectious mononucleosis, however, has been only slight [273], which is perhaps related to the initiation of therapy late during active viral replication. However, acyclovir has been useful in the clinical management of oral hairy leukoplakia [284], and viral shedding has decreased in compromised patients [263, 269].
Ganciclovir and interferon have also been noted to be active against EBV [285–290]. The activity of acyclovir in the treatment of EBV lymphoproliferative syndromes has not been particularly salutary, although at least some individual patients with polyclonal disorders may have benefited [243, 265]. However, in a more comprehensive compilation of cases, there did not appear to be any difference in survival of those receiving acyclovir therapy for EBV-associated lymphomas [267]. More important in the overall management is reduction of intensive immunosuppressive therapy, which can have salutary effects early in the evolution of the lymphoproliferative disorders [239, 259]. Once mutations in oncogenes or tumor suppressor genes have taken place, attempts to reduce immunosuppressive therapy may be futile [250], and cytotoxic therapy may be more appropriate. Soluble CR-2 (the EBV receptor of B lymphocytes) has been proposed as a therapeutic agent that can block EBV infection [291].
In solid organ transplant patients, the number of EBV-infected lymphocytes in the peripheral blood has been noted to correlate with the risk for posttransfusion lymphoproliferative disorder (PTLD) [292]. After BMT, high levels of circulating EBV detected by quantitative PCR have similarly been shown to be associated with the subsequent development of PTLD [293–295]. This affords an early opportunity to intervene “preemptively” by reduction in the immunosuppression if possible.
Rituximab, an anti-CD20 monoclonal antibody, has also been used to treat lymphoproliferative disorders either by itself or in combination with lymphocytes, and in anecdotal reports it has demonstrated efficacy [296–298]. Administration of rituximab can be used preemptively to prevent PTLD in BMT patients with high levels of circulating EBV [299]. In healthy seropositive individuals, the population of EBV-cytotoxic T-cell precursors in the peripheral blood is much higher than populations of cells sensitized to other herpesviruses. Thus, buffy-coat leukocyte infusions or ex vivo expanded EBV-specific cytotoxic lymphocytes can be used as therapeutic modalities for EBV-associated PTLD [300]. Infusions of EBV-specific cytotoxic T lymphocytes have been given to patients with high levels of virally infected lymphocytes at high risk for PTLD, resulting in a reduction of viral load [293, 301–304]. Antiviral agents have not been successful in preventing the development of PTLD [272, 274, 277]. Gene-marked infused cells can persist for as long as 18 months [305]. Such “preemptive” lymphocyte transfusions offer promise for prevention of PTLD in at risk patients.
Human Herpesvirus Types 6, 7, and 8
Human herpesviruses types 6 and 7 have only been recognized in the last decade. However, infection in humans is common [306, 307]. Both have a preference for CD4-positive T lymphocytes but can infect a variety of T- and B-cell lines. Seropositivity is present in 50–90 % of normal adults, and infection generally occurs during the first decade of life. Both appear to share clinical manifestations. Although usually subclinical, exanthem subitum, a self-limited, mild, febrile illness, can occur. Viral antigens to HHV-6 have been noted in tissues from lymphoma, but their significance is as yet unknown [308–310].
Reactivation of HHV-6 is frequently detected in the blood of BMT recipients [311–314]. For the most part this has not been associated with clinical manifestations. However, suggestions have been made that rash [313], GvHD [314, 315], and marrow suppression in the posttransplant setting [316–318] may be associated. Other reports have suggested a role for HHV-6 in interstitial pneumonitis [319, 320], encephalitis [321], and lymphocytopenia [322]. The precise role in pathogenicity of this virus has been debated [323–325].
The true impact of antiviral therapy for the treatment of clinical disease due to reactivation or primary infection with HHV6 remains unknown due to the lack of well-controlled, prospective clinical trials. Antiviral agents including ganciclovir and foscarnet appear to be active against HHV-6 in in vitro assays [326] and have been used anecdotally [327–335].
Much less is known regarding human herpesvirus 7. It has been implicated as a possible cofactor in human herpesvirus 6 infections [335] and has been implicated in a case of acute myelitis in an adult BMT recipient [336].
Human herpesvirus type 8 has been associated with several human malignancies, including Kaposi’s sarcoma (giving rise to its other name, Kaposi’s sarcoma-associated herpesvirus), some forms of lymphoma, and multiple myeloma. This virus has been found in saliva, semen, and peripheral blood mononuclear cells and is thought to be transmitted sexually. Tissue samples from most Kaposi’s sarcomas contain gene sequences in common with HHV-8 [337–339]. Serologic prevalence studies have revealed contradictory findings. Certain forms of B-cell lymphomas have also been described, especially AIDS-associated body cavity lymphomas, as containing HHV-8 sequences [340, 341]. It is thought to be the cause of Casteman’s disease [342]. Several laboratories have reported detection of HHV-8 DNA sequences in patients with multiple myeloma [343]; however, other groups have disputed such claims [344, 345].
Intestinal Viruses
Enteric viruses cause sporadic infections in patients with hematologic malignancies, which generally occur in conjunction with outbreaks within a given community. Common pathogens include coxsackievirus, rotavirus, the Norwalk virus, and other caliciviruses and astroviruses. But other viruses, including adenoviruses, present in the wider community may also cause illness. Coxsackie A1 is an enterovirus that has been reported to cause severe diarrhea in marrow transplant recipients [346]. In one series, episodes of gastroenteritis were clustered temporarily and caused high mortality [346]. At autopsy foamy vascularization of the mucosal epithelium was noted, with patchy sloughing of intestinal mucosal epithelium. This outbreak coincided with diarrheal illness in nonimmunocompromised children in the community from whom coxsackievirus was isolated from stool. Isolated cases of disseminated infection from enteroviruses have been reported [347, 348]. Isolation of the virus from stool is confirmatory of the diagnosis. Coxsackievirus infection must be distinguished from GvHD by intestinal biopsy because the manifestations may be quite similar.
Rotavirus is a double-stranded RNA virus and member of the Reoviridae family. Its target tissue is the small intestinal mucosal epithelium. Fecal–oral transmission is the usual mode of contagion. There is considerable seasonal variation of infections, most occurring between January and April in temperate climates. Rotavirus gastroenteritis can vary in severity from mild, short-lived diarrhea to overwhelming and occasionally fatal gastroenteritis. A cluster of cases has been reported in BMT patients [349]. In another study, frequent stool isolation of rotavirus has been found in autologous and allogeneic marrow transplant recipients with diarrhea [350]. At present there is no specific antiviral therapy; electrolyte and fluid replacement are important adjunctive measures. To date there have not been cases of Norwalk and related viruses reported in patients with hematologic malignancies, probably because of the lack of widely available diagnostic assays. Humoral immunity is felt to be an important host protective response for these viral pathogens. The administration of oral immune globulin or breast milk has been suggested for the treatment of severe rotavirus infection.
Adenovirus
Adenoviruses are double-stranded DNA viruses. Forty-nine antigenic serotypes (divided into six subgroups, A through F, based on their antigenic properties and hemagglutination properties) have been identified, each associated with different clinical syndromes. For example, serotype 3 has been associated with keratoconjunctivitis, serotype 11 with hemorrhagic cystitis [351–353], serotypes 40 and 41 with gastroenteritis, and serotypes 3, 4, and 7 have been documented in epidemics of acute respiratory diseases in military facilities. Serotype 35, although uncommmon in the general population is common in immunocompromised patients.
The means of transmission of adenovirus is uncertain but has been reported to occur via direct contact, aerosols, contact with respiratory or fecal secretions, and contaminated water. The virus is capable of at least three types of interactions with cells. These include a lytic infection, a chronic persistent or latent infection, and oncogenic transformation.
Patterns of Infection
Adenovirus is endemic in the pediatric population, with approximately 80 % of children between the ages of 1 and 5 years having antibody to one or more serotypes [354, 355]. Adenovirus is a relatively common cause of respiratory tract infections in children less than 5 years of age, causing 2–7 % of respiratory tract infections and 5–11 % of viral pneumonia and bronchiolitis cases [356]. These infections are rarely severe and typically manifest as upper respiratory tract infections. Other presentations include keratoconjunctivitis, gastroenteritis, cystitis, myocarditis, and lower respiratory tract infections.
Adenovirus infections are well documented in immunocompromised patients, including BMT recipients (4–21 %), solid organ transplantation patients (liver transplants 8–18 %, renal transplants 12 %), and patients with AIDS (28 %). The frequency with which adenovirus infection and disease is reported among BMT patients is increasing. This is due in part to increased intensity of immunosuppression and to the use of T-cell-depleted cell products. In the mid-1980s, approximately 5 % of all marrow transplant recipients were reported to have adenovirus recovered [351, 357], and that number increased to 12–21 % in the mid-to-late 1990s [358, 359].
In the BMT population, approximately 33 % of infected patients develop severe and prolonged symptoms, such as (1) gastrointestinal infection, manifested as diarrhea, hemorrhagic colitis, or hepatitis; (2) urinary tract infections, manifested as hemorrhagic cystitis often associated with renal failure; and (3) pulmonary infections, resulting in interstitial pneumonia. Adenovirus infections in hematopoietic stem cell transplant (HSCT) recipients are often disseminated, which is defined as isolation of the virus from two different sites or from the blood [349, 356, 358, 360–364].
The BMT programs at both the University of Kentucky and the University of Wisconsin have reviewed the incidence of adenovirus infections in their transplant population [362, 364]. Using different definitions for infection and disease, these programs have established risk factors for adenovirus disease in BMT recipients. First, the age of the BMT recipient is predictive for infection, with pediatric patients more likely to have a positive culture for adenovirus than a similar adult population (23 versus 9 %, p < 0.0001). The reasons for this remain unknown, and the type of transplant procedure does not explain this finding. Second, the type of BMT predicts for infection. The Kentucky program demonstrated that allogeneic BMT recipients are more likely to develop infection as compared to autologous recipients (16 versus 3 %, p < 0.0001) [359]. Differences have been shown in some series among allogeneic patients, depending on the degree of HLA disparity. A higher incidence of both adenovirus infection and disease was seen in patients undergoing unrelated HLA-mismatched grafts compared with related HLA-matched grafts, although the difference was not statistically significant [354, 357]. Finally, adenovirus disease developed in a greater number of patients with moderate-to-severe GvHD (15 versus 5 p < 0.05). The development of moderate-to-severe GvHD was found to be an independent risk factor predictive for adenovirus disease on multivariate analysis (p < 0.01; odds ratio = 5.4; 95 % confidence interval 1.6–17.9). The other major risk factor for progression of infection to disease is isolation of adenovirus from multiple sites. Isolation of adenovirus from more than two sites strongly correlates with an increased risk of invasive disease compared to isolation from one site; adenovirus disease occurred in 63 % of patients with more than two positive sites compared to 11 % with only one positive site.
The development of invasive disease is associated with a shorter median survival. The median survival for all patients who had a positive adenovirus culture was 208 days. Patients with evidence of invasive infection had a median survival of 100 days, compared to 998 days for those with noninvasive adenovirus infection [364]. Of those patients who develop invasive infection, 50 % die.
Treatment
Clearance of adenovirus has been shown to correlate with recovery of adenovirus-specific T-cell immunity [365, 366]. When possible, rapid tapering or withdrawal of immunosuppression is felt to be the best way to prevent progression of adenoviral infection [367–370]. Currently, there is no reliably effective antiviral therapy for the treatment of adenovirus infections. There are numerous case reports in the literature reporting success with ganciclovir [371–373], intravenous ribavirin [374–381], vidarabine [382, 383], and cidofovir [384–386]. The key to successful treatment is prompt initiation of therapy in high-risk individuals. A risk-adapted approach, similar to that taken with CMV, may in fact prevent progression to disseminated disease, which is typically associated with a dismal outcome. An alternative strategy that has been exploited is adoptive immunotherapy. There are data supporting the use of donor leukocytes to treat posttransplant EBV-associated lymphoproliferative disease, as discussed previously. This principle has been extrapolated to the treatment of life-threatening adenoviral infection following T-cell-depleted HSCT and has met with success, although there are only published data on one patient [387].
Clearly there is an absence of adequately controlled studies on treatment of adenovirus infections with the currently available antiviral agents. Cidofovir and ribavirin appear to offer most promise and should be further investigated in a prospective multicenter trial.
Prevention
In the allogeneic transplant population, patients can be stratified according to their risk of adenoviral disease:
Intermediate. T-cell replete, related-donor allograft recipients without GVHD
High. T-cell depleted (2–3 log) related or unrelated-donor transplants [374, 375]; HLA-mismatched transplants (other than DRB1 mismatch) [391]; patients with GVHD on systemic steroids [360, 361, 390], pediatric patients
Highest. refractory GVHD; unrelated cord blood or haploidentical stem cell graft, T-cell depleted (>2–3 log), use of anti-T-cell antibodies (antithymocyte antibody, alemtuzumab) [374, 392]
It is currently advised that patients in the highest risk category undergo weekly PCR monitoring for active adenovirus for either the first 6 months after transplant or for the duration of immunosuppression [391, 393]. Unfortunately, there is no definitive data on a critical value for viral load in the peripheral blood to indicate initiation of treatment.
Respiratory Viruses
Respiratory viral infections are a common cause of morbidity and mortality amongst patients with hematological malignancies. Despite significant advancements that have been made in our understanding of the pathophysiology, epidemiology, and laboratory diagnosis of these infections; improvement in available treatment regimens for established disease has been limited. A major focus has therefore remained on prevention. Along with adenovirus (already discussed previously) which can also present as an upper respiratory infection, respiratory syncytial virus, influenza, and parainfluenza are the most commonly encountered and will be discussed next [394–396].
Respiratory Syncytial Virus
Respiratory syncytial virus (RSV), a member of the paramyxoviruses, is a single-stranded RNA virus related to parainfluenza virus and is a well-recognized cause of upper and lower respiratory tract illness in children [397–400]. Outbreaks of RSV infection are seasonal and usually occur in the community during November to April, the fall and winter months in the Northern Hemisphere [401, 402]. Repeated infections are common in all age groups, and previous infection does not confer immunity and prevent subsequent infections, even in sequential years [403, 404].
The virus can be transmitted by three possible mechanisms [405]. The first is transmission by small-particle (less than 10 μm diameter) aerosols, which are typically produced by coughing or sneezing. These are able to traverse distances of 1.8 m or greater. Second is transmission by droplets or large particles. This type of transmission necessitates person-to-person contact, usually at distances of less than 0.9 m for infection to occur. Finally, transmission can occur via contact with contaminated surfaces. Typical winter conditions enable RSV to remain viable on nonporous surfaces for longer than 6–12 h. Therefore, assuming virus viability, there is great potential for self-inoculation and transmission of infection to surrounding patients and medical personnel. This is particularly worrisome for medical personnel and nursing staff, who may unwittingly spread the virus during high-risk months. Prevention using good infection control practices is of paramount importance. Nosocomial transmission has been implicated in some groups of immunocompromised patients [406]. Respiratory secretions are contagious and the portal of entry is through the nasopharyngeal mucosa and conjunctivae. The incubation period of RSV is 3–5 days. The target tissue is the respiratory epithelium. Proliferation and necrosis of bronchiolar epithelium with lymphocytic peribronchiolar infiltration and edema are characteristic pathologic findings. Both humoral and cell-mediated immunity are important protective responses [407], and those individuals with inherited or acquired immunodeficiency have more severe and long-lasting RSV infections than normal individuals [400].
RSV infections in BMT patients have been increasingly described since 1988, with several outbreaks being reported in large BMT centers [401–410]. The reported incidence of RSV infections in hospitalized BMT patients ranges from 7 to 20 %. High-risk groups for the development of RSV infections include premature infants; children with bronchopulmonary dysplasia, congenital heart disease, or cystic fibrosis; immunosuppressed patients, including bone marrow and solid organ transplant recipients and those with underlying disorders of cellular immunity; elderly people; and those living in institutions [401]. Children with hematologic malignancies are susceptible to RSV infection, which has the potential to cause more severe illness in these patients than in nonimmunocompromised patients [399, 408], and can result in extension to the lower respiratory tract, pneumonia, and death. Adults with hematologic malignancy are also susceptible [409, 410], especially those profoundly immunosuppressed after marrow transplantation.
In immunocompromised children and adults, RSV infections usually present as an upper respiratory tract illness with cough, fever, sinus and nasal congestion, rhinorrhea, and dyspnea but rapidly progress to severe and often fatal lower respiratory tract viral pneumonia, with marked radiographic changes [411]. A review of 600 children with RSV infection revealed 47 patients with compromised immune function over a 10-year time span. On analysis of the data, those children with primary immunodeficiency disorders and those receiving chemotherapy were more likely to develop RSV pneumonia (60–80 % admitted to intensive care units) and had a higher mortality rate (15–40 %) compared with those with normal immunity (5 % intensive care unit admissions; fewer than 1 % died) [399]. In the BMT population, lower respiratory tract pneumonia develops in approximately 50–60 % of RSV-infected patients [402, 411]. The development of pneumonia is associated with a higher mortality rate (66–100 %), and therefore rapid treatment and prevention of progression are important [401, 409, 412, 413]. The M.D. Anderson Cancer Center has produced data demonstrating that those patients treated earlier (1 day or less from symptoms) had an overall mortality rate of 22 % versus 100 % in patients who had delayed treatment or who did not receive treatment [409]. This confirms that the mortality rate increases as the time to diagnosis and treatment increases. The other known predictor of outcome in BMT recipients is engraftment status. The risk of pneumonia is greater in patients who have not engrafted than in those who have (79 versus 41 %) [411]. There are also data demonstrating that the frequency of progression from an upper respiratory tract infection to pneumonia is higher in patients in whom engraftment has not occurred or who are less than 1 month posttransplant, compared to those who are more than 1 month posttransplant/engraftment (70–80 to% versus 25–40 %, respectively). However, once pneumonia develops, engraftment status does not alter overall mortality [402, 412]. Other risk factors that have been identified as causing progression of RSV infections from an upper respiratory tract to a lower respiratory tract infection include older age and HLA-mismatched or unrelated donor transplants (relative risk 2.8.1) [414].
Diagnosis
Nasopharyngeal aspirates or washes are among the most common specimens used for the diagnosis of RSV. Wash specimens are preferred to swabs because they have increased sensitivity. Diagnosis can be made by molecular and nonmolecular methods. Specimens may be tested for RSV by culture or by use of a rapid detection assay such as enzyme-linked immunoabsorbent assay (ELISA) or immunofluorescence [400]. More sensitive methods such as PCR analysis for the presence of viral specific nucleic acid have been more utilized with more recent attention to the quantitation of viral load. Specimens from sputum or bronchoalveolar lavage also contain the pathogen, and in the immunocompromised patient, bronchoalveolar lavage has an increased sensitivity for virus detection
Treatment
Currently there are a number of available treatment options for RSV pneumonia which include ribavirin, intravenous immunoglobulin rich in RSV titers, RSV immune globulin (RSV-IG [Respigam, MedImmune, Gaithersburg, MD]), and palivizumab (Synagis, [MedImmune, Gaithersburg, MD]). Unfortunately there are no large randomized controlled trials in patients with hematological malignancy supporting the efficacy of any one of these treatments alone or in combination. The most commonly employed agent, ribavirin is a synthetic purine nucleoside derivative [415]. Its antiviral effects result from three mechanisms of viral inhibition, namely, suppression of viral nucleic acid synthesis; blocking of the formation of the terminus of mRNA, leading to inefficient translation of mRNA into viral structural proteins; and suppression of viral polymerase, resulting in interference with viral mRNA expression and subsequent protein synthesis. Ribavirin has activity against a broad spectrum of both RNA and DNA viruses and has been investigated as a potential treatment in influenza A and B, disseminated adenovirus, parainfluenza virus, herpesvirus, and RSV infections. Ribavarin may be given orally (15–20 mg/kg/day divided into 2–3 doses) or aerosolized (6 g administered continuously or 2 g three times per day). Its role in RSV infections is controversial with much of the data on treating RSV infections come from the pediatric literature, as this is one of the populations most at risk for this infection. Conflicting results are reported on the use of ribavirin to treat RSV infections in infants and children. Several studies of infants and children on ventilators have examined the effect of ribavirin on days of ventilation and hospitalization. The results are variable, with one study showing a reduction in days of ventilation and hospitalization [416], one study demonstrating no difference in hospitalization [417], and two studies showing prolonged hospitalization and ventilation compared to placebo [418, 419]. A meta-analysis of studies in infants with RSV lower respiratory tract infections demonstrated an overall trend in favor of ribavirin but no evidence of a significant benefit; unfortunately, the studies lacked the power to detect reductions in mortality [420]. Based on these data, ribavirin is not often used for infants and children with RSV infections and is reserved for immunocompromised and critically ill patients.
An abundance of case reports and small series report the effectiveness of aerosolized ribavirin in the treatment of RSV in both adult and pediatric BMT recipients [401, 409, 421–424]. It is important to remember that the number of patients treated is small and the studies are uncontrolled and noncomparative. One of the largest studies of RSV infection in BMT recipients reported 31 cases of RSV infection diagnosed among 199 transplant recipients [401]. Of the 31 patients, 18 were diagnosed with RSV pneumonia and the remaining 13 had upper respiratory tract infections. Ribavirin was administered to 13 (72 %) of the patients with pneumonia but to none of the patients with upper respiratory tract infections. Survival among patients who developed pneumonia was poor, with a mortality rate of 78 %. The mortality rate was 70 % in the 13 patients treated with aerosolized ribavirin monotherapy and 100 % in the untreated patients. Responses appeared to be better in those patients receiving more than 5 days of ribavirin therapy. An earlier study also documented a mortality rate of 66 % in BMT patients with radiographically documented RSV pneumonia treated with aerosolized ribavirin monotherapy [413]. Based on these findings and the results outlined in case reports in the literature, it is difficult to justify monotherapy with ribavirin for the treatment of RSV pneumonia in immunocompromised individuals. If ribavirin is to be effective, early diagnosis is essential and treatment should be instituted prior to progression to pneumonia. In a randomized controlled trial, the efficacy and tolerability of aerosolized ribavirin for preventing the progression of RSV upper respiratory tract infection (URI) to a lower respiratory tract infection (LRTI) was studied in 14 patients who had undergone myeloablative conditioning and were 90 days post-HSCT [425]. Patients were considered to be infected if they had upper respiratory signs, respiratory rate greater than 150 % of baseline, increasing cough, wheezing, sputum production, or pleuritic chest pain and oxygen saturation less than 90 % on 2 consecutive measurements and a documented RSV infection. Patients randomized to drug therapy received aerosolized ribavirin 2 g (over 2 h) three times each day for 10 days. The primary outcome was progression to clinical pneumonia. Following randomization, 9 patients were allocated to treatment and 5 patients received supportive care. Clinical pneumonia after randomization occurred in 1/9 (11.1 %) ribavirin patients and 2/5 (40 %) of control patients (p = 0.51). There were no deaths in the trial. Overall, this study did not reveal any differences in outcomes despite trends in lower viral loads among ribavirin recipients.
In a study by Chemaly et al., the effect of aerosolized ribavirin on preventing pneumonia in 107 HSCT or hematologic malignancy patients with RSV upper respiratory infection was reported [426]. Patients received 6 grams of aerosolized ribavirin through a face mask inside a scavenging tent for 18 h/day (duration of therapy not defined). In this study, the authors identified 107 RSV patients, 61 were treated with ribavirin and 46 were provided supportive care. Progression to pneumonia occurred in 12/61 (20 %) and 27/46 (59 %) of patients, respectively (p < 0.005). On multivariate analysis, patients who developed pneumonia were older (OR, 1.037; 95 % CI, 1.001–1.074; p = 0.042) and did not receive RSV-directed antiviral therapy (OR, 4.67; 95 % CI, 1.20–18.10; p = 0.025). In addition, degree of lymphopenia (absolute lymphocyte count <200 cells/ml) was associated with poor outcomes and should be included in the treatment algorithm.
A number of small studies have suggested improved response rates and improved survival rates with combination therapy [427]. Whimbey et al. [428]. studied aerosolized ribavirin 20 mg/ml for 18 h/day in combination with IVIG 500 mg/kg every other day in BMT patients who were RSV positive. The IVIG product was chosen based on the presence of neutralizing antibodies against RSV subtypes A and B. Of 42 patients who presented with upper respiratory tract illness, 19 were RSV positive. Pneumonia developed in 16 patients, of whom 12 (75 %) were treated with combination therapy. The overall mortality rate among those with pneumonia was 42 %, a significant improvement over mortality rates associated with ribavirin monotherapy. As seen with ribavirin monotherapy, responses were more likely to occur with fast implementation of treatment. The same group from Texas used the same combination of drugs (ribavirin plus IVIG) in a trial evaluating efficacy in preventing progression of RSV upper respiratory tract infection to pneumonia [412]. Fourteen patients were treated for a mean of 13 days with combination therapy. The upper respiratory illness resolved in 10 patients (71 %). Four patients (29 %) developed pneumonia, which was fatal in two. In this study, two different methods of administering ribavirin were evaluated. Traditionally, aerosolized ribavirin is administered at a daily dose of 6 g, delivered at a concentration of 20 mg/ml for 18 h/day in a scavenging tent to prevent environmental contamination with the drug [412]. Although it produces very high drug levels in bronchial secretions with little systemic absorption [429, 430], this method of administration is cumbersome and is often difficult for patients to tolerate for long periods. In an attempt to improve compliance and ease of administration, short-duration aerosolized ribavirin at a daily dose of 6 g, administered at a concentration of 60 mg/ml for 2 h every 8 h was evaluated. This method of administration has been attempted in the past with limited success [411], but the favorable results reported by the Texas group offer promise and should be further evaluated in a larger group of patients.
The doses of standard IVIG necessary to achieve the required in vivo RSV-neutralizing activity are large, and frequent dosing is required. This, coupled with wide variations in RSV antibodies between batches and commercial sources, led to the development of a commercial RSV hyperimmune globulin. RSV-IG, a polyclonal hyperimmune globulin containing high titers of RSV antibodies (over 19,200 MU/ml) against RSV [431]. The product is commercially available as Respigam (MedImmune, Gaithersburg, MD).
The only available data in immunocompromised patients are in the BMT literature. The results of a compassionate use protocol of RSV-IG in the treatment of lower respiratory tract RSV infections in pediatric BMT patients showed that 55 % of patients had resolution of lower respiratory tract infections following one dose [431]. Importantly it must be noted that of the patients enrolled in the program, 82 % had previously received aerosolized ribavirin and 91 % were receiving concurrent aerosolized ribavirin with RSV-IG. Despite concomitant treatment, the mortality rate was only 9 %, and this offers promise for the future. Clearly, further studies using RSV-IG as monotherapy and comparing outcomes to those obtained with ribavirin are warranted in the immunocompromised patient population.
In addition to RSV-IG, a humanized RSV monoclonal IgG1 monoclonal antibody that is highly active against RSV types A and B is commercially available [432]. Palivizumab is approved by the US Food and Drug Administration for the prevention of serious RSV lower respiratory tract disease in pediatric patients at high risk for RSV disease. In the pediatric population it has been shown to reduce hospitalizations as a result of RSV infection by 55 %, reduce the total number of days in hospital as a result of RSV infection (36 versus 63 days), reduce the number of ICU admissions (1.3 versus 3 %), and reduce the total number of days hospitalized overall (191 versus 242 days) [433]. In view of these promising data and the increasing incidence of RSV infection among immunocompromised patients, the use of palivizumab has been evaluated in a small number of BMT patients. There are two small studies in this patient group, one examining the pharmacokinetic profile of palivizumab in patients without RSV infection, and the other in patients with documented RSV infection. In the pharmacokinetic study, the mean serum concentration after 30 days was 41.9 μg/ml [434], which was within the limits necessary to produce a two-log (99 %) reduction in pulmonary RSV infection, based on animal data [435]. Patients were also monitored for the development of antipalivizumab antibodies. No antibodies were detected, which is consistent with the pediatric literature [434]. The second study evaluated the response to palivizumab in 15 patients with proven RSV infections. At enrollment, 12 patients (80 %) had lower respiratory tract involvement and 3 (20 %) received mechanical ventilation. Palivizumab 15 mg/kg was administered in combination with ribavirin therapy. The survival rate was 87 %, similar to results seen with RSV-IG. Palivizumab is an interesting addition to the therapeutic armamentarium against RSV and offers the advantage of being administered once and producing high concentrations that are sustainable and protective over a 30-day dosing interval. To date there are no comparative studies of RSVIG and palivizumab, so which one is superior remains to be seen.
McCoy et al. recently reported their single-center experience with a RSV algorithm developed by interdisciplinary team for managing RSV infections in 26 patients with hematologic malignancies [436]. Practice guidelines were used to stratify patients by disease severity, time post-HSCT, and degree of lymphopenia. Patients with URI and less than 1 month post-HSCT and/or absolute lymphocyte count <300 cells/mm3 received aerosolized ribavirin (6 g over 12 h for at least 3 days). If patients presented with LRTI, ribavirin and palivizumab (15 mg/kg for one (1) dose) were administered. Upon review of the data, 13 patients presented with URI and 13 patients presented with LRTI. The average number of ribavirin doses was 5 ± 2.1. No mortality occurred within 30 days of RSV diagnosis. No other variables were addressed regarding the efficacy of the regimen.
Khanna et al. also published the experience from their center along with a review of the literature focusing on the treatment of RSV infections in patients with hematological diseases [437]. The authors of this study identified 780 RSV-infected patients from 1981 through 2007. Aerosolized ribavirin was administered to 337 patients with URI and LRTI. In patients with URI, 30/95 (32 %) and 76/112 (68 %) progressed to LRTI for ribavirin treated and untreated patients, respectively. In patients with LRTI, death occurred for 39/102 (38 %) of patients treated with aerosolized ribavirin, 50/116 (43 %) treated with combination ribavirin and IVIG, 15/36 (42 %) of untreated patients. These data suggest that early treatment may play a role in reducing progression to LRTI and that treatment may have minimal benefit once disease has progressed beyond URI. RSV infections are not only amenable to aerosolized therapy but may be treated with oral therapy. Oral ribavirin is rapidly absorbed and has been evaluated in several reports. Khanna et al. performed a retrospective analysis on the efficacy of oral ribavirin ± immunoglobulin (IVIG) and/or palivizumab antiviral agents in 34 patients with RSV infection and hematological diseases. Patients were stratified based on degree of immunosuppression and the presence of URI or LRTI. Patients were stratified into moderate or severe immunosuppression based on time from transplant, time from T or B cell depletion, presence of lymphopenia, or GvHD grade ≥2. Patients with moderate immunosuppression received oral ribavirin (1,200–1,800 mg/day) plus IVIG (once a week). Patients with severe immunosuppression with URI or LRTI received oral ribavirin plus IVIG plus palivizumab. At the time of RSV diagnosis, 22/34 had URI and 12/34 had LRTI. In patients with URI, 10/22 had moderate immunosuppression and none of the 10 progressed to LRTI. In 12 patients with severe immunosuppression and URI, 2/12 progressed to LRTI. In patients with LRTI and severe immunosuppression, five (5) out of 10 died. For those patients who died, degree of immunosuppression and the presence of LRTI were associated with a poor outcome. RSV loads in nasal secretions were evaluated in 19 patients. A greater than 2 log10 copies/ml drop from baseline was seen in 11/19 within 7 days. None of these patients went on to develop LRTI. Overall, oral antiviral therapy was most effective in preventing disease progression in those patients with moderate immunosuppression and URI.
Avetisyan et al. evaluated the role of systemic ribavirin (intravenous or oral at 15–20 mg/kg/day) therapy in 28 HSCT recipients with laboratory verified RSV infection [438]. No adjunctive therapy was administered (e.g., palivizumab). Following treatment, all patients with URI did not progress to LRTI and survived. For patients with LRTI, 5/14 died directly from RSV. Overall mortality was approximately 20 % most of which was in those patients with LRTI. Of note, these data are similar to other reports in which broader antiviral therapy was applied. Overall, these data speak to the fact that degree of immunosuppression and presence of LRTI are associated with poor outcomes and may not be amenable to combination therapy.
With mortality rates approaching 40–60 % in patients with lower respiratory infection, ribavirin has become an option for treating patients with upper respiratory disease in the hope of minimizing disease progression. Currently available data suggest that early recognition of infection with RSV, supportive care measures, such as oxygenation and hydration, and reduction of immunosuppressive therapy if possible may be beneficial in improving outcome in improving outcomes. Antiviral therapy appears to be most effective when it is started within 48–72 h of symptom onset, although there are no large randomized controlled trials available to help determine the optimal treatment regimen. With low-level evidence, most data suggest that ribavirin is probably beneficial in preventing disease progression in patients with symptoms of upper respiratory infection. Unfortunately, the initiation of antiviral therapy once lower respiratory infection has developed has not shown a significant benefit in reducing the risk of RSV-related mortality with or without an additional immunotherapy in the form of IVIG or monoclonal antibody.
Although it appears that patients undergoing therapy for hematological malignancy with upper respiratory infection with RSH may benefit from treatment with ribavirin by preventing disease progression, a number of questions regarding treatment remain. One such question is the appropriate route of administration of the drug, particularly in light of the challenges in delivery of the aerosolized formulation of ribivarin. In the US, aerosolized ribavirin is the only FDA-approved agent available for the treatment of RSV approved in 1985. Therefore, most early studies in patients with hematologic malignancies utilized this form of the drug. Since then, aerosolized administration optimized drug concentrations at the site of infection interest stayed with the aerosolized product. There are reasons, however, to consider that oral ribavirin could replace aerosolized therapy in managing respiratory viruses in patients with hematological malignancy. Ribavirin is rapidly absorbed with a reasonable bioavailability at 64 % (extensive first pass metabolism), has a large volume of distribution due to tissue and intracellular penetration, and has data to support its role in reducing disease progression (similar to inhaled). In addition, oral therapy is safer for healthcare workers (avoids environmental exposure) and significantly less expensive (Avetisyan). Well-controlled clinical trials to better define the appropriate antiviral management of infection with RSV in this patient population are warranted.
Prevention
Patients presenting for BMT during the RSV season with respiratory symptoms should receive workups for conventional respiratory viruses (CRVs), including RSV, adenovirus, and influenza and parainfluenza viruses. If any of these viruses are isolated, the BMT should be placed on hold until resolution of the acute process if possible. Similarly, patients with cancer undergoing chemotherapy should have therapy temporarily suspended if possible [134].
If any hospitalized BMT patient develops signs and symptoms of CRV infection while an inpatient, appropriate specimens should be obtained for viral culture and rapid diagnostic tests for RSV [439]. If the patient continues to display respiratory signs and rapid testing is negative (two diagnostic samples more than 2 days apart), a bronchoalveolar lavage should be considered. These patients should be placed under contact precautions to prevent spread of infection.
Health care workers (HCWs) handling respiratory symptoms of infected patients should wear gowns, surgical masks, and eye protection to avoid contamination from secretions. Protective clothing should be put on when entering the patient’s room and discarded before leaving the room. HCWs should change gloves between contacts with patients. All visitors of an infected patient should wash their hands after contacts with the patient or with secretions from the patient. In addition, visitors with upper respiratory tract infections should defer visiting until resolution of symptoms, and HCWs with respiratory symptoms should be reassigned to duties other than patient care. Unfortunately clinical trials with inactivated and attenuated vaccines have not proven benefit. Further study of subunit vaccines are under development.
Influenza Viruses
Influenza A and B viruses are members of the Orthomyxoviridae family. These enveloped single-stranded RNA viruses are important causes of recurrent epidemics of respiratory disease in both normal and immunocompromised patients. Four genera exist. influenzavirus A, influenzavirus B, influenzavirus C, and thogotovirus [440]. Influenza A viruses are further divided into subtypes on the basis of serological and genetic differences in their surface glucoproteins [440]. Fifteen subtypes of hemagglutinin (H1 to H15) and nine subtypes of neuraminidase (N1 to N9) have been identified. Considerable antigenic variations can occur; this property has made control by vaccine difficult because these antigenic changes can permit the organism to evade established immunity. Influenza viruses are named by type, location of isolation, number of isolates, year of recovery, and for influenza A, the subtype (e.g., A/Texas/36/91[H1N1]) [441]. Three influenza A virus subtypes, H1N1, H2N2, and H3N2, have caused extensive human diseases during the last century.
Infected respiratory secretions are generally the mode of transmission to a susceptible individual. The target cells are those of the respiratory tract epithelium, particularly the columnar epithelial cells, where the influenza virus replicates [441]. Desquamation of epithelial cells, sloughing and in more severe cases, hemorrhage, hyaline membrane formation, and neutrophilic infiltration are notable findings pathologically. Influenza typically presents clinically as fever with associated chills. Other common presenting symptoms include sore throat, headache, myalgias, and fatigue. In more severe cases patients may present with fulminant pneumonia, a common presentation in the elderly in influenza season. The incubation period for influenza viruses is between 1 and 4 days. Secondary bacterial infections (mainly Streptococcus pneumoniae, Staphylococcus aureus, and Haemophilus influenzae) are well-recognized complications. Humoral immunity is an important protective host response, especially neutralizing antibody.
Influenza infections in immunocompromised patients occur, concomitant with outbreaks in the community. For example, an influenza A epidemic in 1991–1992 in Houston was evaluated [442]. Among adult BMT patients, approximately 40 % developed acute respiratory illness, and influenza A was isolated from 30 %. All had upper respiratory tract illness, but this was complicated by pneumonia in 75 %. The case fatality was 17 %. Nosocomial acquisition of influenza is also common in hospitalized adults with leukemia or those undergoing BMT [442–444]. Influenza infections in immunocompromised patients have been studied regularly over the past decade and a review of CRVs recovered from 785 consecutive immunocompromised patients with suspected respiratory tract infections has been published [445]. Diagnosis of CRVs was made following bronchoalveolar lavage in all cases, and 199 viruses were isolated from these specimens (23 %). Influenza virus was isolated in 11 of 37 patients with CRVs and was the most common CRV isolated. Pneumonia was present in 82 % of these patients, and the overall mortality rate was 27 %. These results are similar to those previously published in the literature [410, 446] and indicate the need for stringent preventive measures during influenza season and community outbreaks.
In April 2009, an outbreak of a novel H1N1 influenza A virus was noted in Mexico which had a reportedly high mortality rate in an otherwise “low-risk” patient population [447]. This virus was noted to be a reassortment of DNA including genetic components of Swine and human influenza [448]. As opposed to the seasonal influenza virus, mortality from the newly designated Influenza A/pandemic 2009/H1N1 appeared to be higher in infants, young adults, those with obesity, and pregnant women. It was postulated that older individuals may have had some cross reactive immunity to prior exposure of to strains of influenza A not shared by younger individuals [449]. Shortly after infection with this virus was noted in the United States [450], and two cases of infection with the novel H1N1 (“Swine flu”) virus in allogeneic hematopoietic stem cell transplantation recipients were reported [451]. In those cases, prolonged viral shedding with eventual development of resistance to oseltamivir was noted. One patient improved without change in antiviral therapy and one was treated with zanamivir and ribavarin with persistent illness and prolonged hospitalization. Additional cases have been reported in patients with hematological malignancy and/or undergoing hematopoietic stem cell transplantation with varying outcomes [452]. Although pandemic in its spread across the globe, significant increase in influenza-related mortality like that seen in the 1918 outbreak was not noted; however, this outbreak was a warning to healthcare officials and the general public as to how rapidly a contagion can spread globally.
Detection
There are four broad potential targets in diagnosing influenza [453]: virus isolation, detection of viral proteins, detection of viral nucleic acid, and serological diagnosis. The gold standard diagnostic procedure for influenza is isolation in cell culture. This procedure requires 2–4 days to produce a result. The shell vial technique is much faster, enabling detection of influenza antigens following overnight culture, although the sensitivity of this test is only approximately 80 % as compared to standard cell culture [453]. A new area of emerging diagnostics is the detection of viral proteins. There are several rapid tests now on the market, which are easy to perform and do not require skilled technicians. Point-of-care rapid diagnostic tests that are currently available or under development include the Directigen Flu A test, which detects only influenza A in nasopharyngeal, nasal, or throat specimens. This is an enzyme immunoassay test and can be completed in eight steps in approximately 15 min. The sensitivity of the test is variable, ranging from 50 to 99 %, with a specificity of 52–97 % [440, 441]. The other tests are the ZstatFlu, FLU OIA, and QuickVue. Each of the three latter tests can detect both influenza A and B. The ZstatFlu is a colorimetric neuraminidase enzyme assay that has only three steps but takes 12 min to produce a result from a throat specimen. It has a sensitivity of 62 % and a specificity of 99 %. The FLU OIA assay method is an optical enzyme immunoassay, which can be completed in seven steps. It is rapid, with a 17-min turnaround, a sensitivity of 62–88 %, and a specificity of 70–80 %. The QuickVue takes the longest of all the new techniques; it is able to produce a result from a nasopharyngeal or nasal specimen in 30 min and has a sensitivity of 73–82 % and a specificity of 96–99 %. These tests are rapid and provide quick answers to assist therapeutic decisions, although they are not as sensitive as culture or PCR techniques. In addition, these tests have not been evaluated in the immunocompromised population and the sensitivity and specificity parameters listed are based on studies performed in healthy children and adults. Although these rapid diagnostic tests assist in early treatment, they will not replace the need for viral culture because only culture isolates can provide specific information on circulating influenza subtypes and strains [454]. This information is necessary annually to compare circulating influenza strains and vaccine strains and to formulate vaccines for the coming year.
PCR techniques detect viral nucleic acid in a very sensitive manner, and results can be generated within 24 h. This diagnostic tool is best used in outbreak situations, in which molecular typing and analysis of samples is necessary. Finally, serological diagnosis is based on the detection of a fourfold or greater increase in antibody titers. The test requires two samples, one collected at the onset of symptoms and another 14 days later, and this limits its usefulness in guiding treatment [410]. This test is not as reliable in the immunocompromised population.
Treatment
Amantadine and rimantadine are agents that inhibit an early stage of viral replication and appear to be useful in the prevention and treatment of influenza A in young adults, children, families, and the elderly at risk for exposure [455–459]. Both these drugs act to inhibit the uncoating of the influenza A virus (no activity against influenza B virus) by blocking the activity of the viral M2 protein [454]. Both agents are similarly efficacious (70–90 %), although rimantadine may be associated with fewer side effects [460]. These compounds exert maximal benefit when administered within 48 h of the onset of influenza symptoms. They have been shown to reduce the duration and severity of symptoms when administered in this way, although they have not been shown to prevent serious influenza-related complications such as bacterial or viral pneumonia or exacerbation of chronic diseases. One of the main concerns with the use of amantadine and rimantadine is the emergence of drug resistance. Resistance to these agents has been well documented in the laboratory and in humans [461–464], but the frequency and importance of drug-resistant influenza in immunocompromised patients was uncharacterized until recently. It has since been shown to occur in approximately 30 % of severely immunocompromised patients with influenza and in 83 % of patients with symptomatic disease who shed virus for more than 3 days [461]. In patients who do not respond to treatment, resistant strains must be suspected. Toxicity is also a concern with this class of agents.
A second class of antiviral drugs, the neuraminidase inhibitors, is also available for the treatment of influenza A and B [465–469]. Zanamivir and oseltamivir are both commercially available and FDA approved for the treatment of influenza A and B. They are oral agents and are better tolerated than amantadine and rimantadine. They are not approved for, nor are there currently sufficient data to support their use in the prophylactic setting. Despite this lack of data, in time of community or nosocomial outbreaks of influenza B infections it may be prudent to prophylactically administer these agents to high-risk individuals; this approach certainly deserves further study. These drugs act by selective inhibition of viral neuraminidase. Neuraminidase, essential for viral replication in vitro, promotes the release of progeny virus from infected cells [465]. By inhibiting neuraminidase, zanamivir and oseltamivir block the active site of neuraminidase and leave uncleaved sialic acid residues on the surface of host cells and influenza viral envelopes [455]. Viral hemagglutinin binds to the uncleaved sialic acid residues, resulting in viral aggregation and a reduction in the amount of virus released [454].
Zanamivir is available for use by inhalation only, using the Diskhaler provided in the package. It is approved for the treatment of influenza in patients 12 years and older who have had symptoms for no longer than 2 days. The recommended dose is two inhalations (10-mg dose) twice daily for 5 days. Following inhalation, approximately 7–21 % of the administered dose reaches the lungs, with 70–87 % of the dose being deposited in the oropharynx [454, 466]. A small amount of the drug is absorbed systemically (4–17 %). Zanamivir has an advantage in that the dose does not need to be modified in renal impairment. Oseltamivir is available as an oral capsule. It is approved for the treatment of influenza A and B virus in adults 18 years of age or older within 2 days of the onset of influenza symptoms. The recommended dose for adults is 75 mg by mouth twice daily for 5 days. Doses need to be reduced in patients with creatinine clearances less than 30 ml/min to 75 mg daily. Approximately 80 % of a dose is systemically absorbed [454]. Both of the commercially available neuraminidase inhibitors have favorable side effect profiles. The most common side effects seen include nausea, sinusitis, nasal signs and symptoms, diarrhea, cough, dizziness, and headaches. The majority of these side effects could be attributed to the underlying influenza infection and were seen with a similar incidence in the placebo groups of the earlier trials. Both neuraminidase inhibitors have been shown in clinical trials in healthy volunteers to reduce the symptoms associated with influenza by approximately 1 day. There is currently a lack of data supporting the use of these agents in immunocompromised patients, patients who are hospitalized, or patients with severe influenza including pneumonia.
As seen with amantadine and rimantadine, resistance to neuraminidase inhibitors can be induced. The emergence of resistance, however, is much less likely to happen, requiring several passages in cell culture [454], whereas resistance to amantadine and rimantadine requires far fewer passages in cell culture. Resistance to zanamivir has already been documented in a BMT recipient [470], and therefore the use of these agents in this population warrants close surveillance. In addition, osteltamivir-resistant novel influenza H1N1 virus infection has also been reported during the recent outbreak [451].
Machod et al. described the benefit of early administration of antiviral therapy in HSCT recipients with the administration of oseltamivir within 48 h of initial symptoms likely to prevent more serious disease [471]. The optimal duration of therapy for patients who develop influenza infections, however, remains unanswered. Prolonged viral shedding leads to the potential of development of drug resistance. In a report by Khanna et al., 18 of 21 episodes of influenza in allogeneic transplant recipients were treated with 75 mg of oseltamivir twice daily for a median of 11 days (8–14) and all survived [472]. Recurrent viral shedding and need for retreatment has also been described [473].
Prevention
Previously, amantadine and rimantadine were used primarily as chemoprophylaxis for community and nosocomial outbreaks of influenza A. Their use has largely been supplanted by the neuraminidase inhibitors. It is currently advised that during community or nosocomial outbreaks, the following stem cell transplant populations should receive chemoprophylaxis with a neuraminidase inhibitor [134]:
1.
Transplant recipients less than 6 months after transplant.
2.
Transplant recipients who have not received a current influenza vaccination and are greater than 4 months after transplantation.
3.
Transplant recipients who are less than 24 months after transplant or who are greater than 24 months after transplant and substantially immunocompromised who have been exposed to influenza. Additionally caregivers and close contacts of transplant recipients should also receive chemoprophylaxis if an outbreak occurs with a strain of influenza not contained in the available influenza vaccine [134].
Inactivated vaccines are available, and community immunization programs in the fall and early winter can reduce the spread within communities. The efficacy of the influenza vaccine in cancer patients and the severely immunocompromised host is reduced as compared to normal controls. Several attempts to overcome poor immune responses, including administering additional injections, have been made although they have not met with much success. Patients with mild underlying immunodeficiency have been able to mount an antibody response. The scenario in BMT patients is somewhat different. In an evaluation of antibody responses to a two-dose regimen of influenza vaccine in allogeneic T-cell-depleted and autologous BMT patients, the likelihood of response was significantly related to a longer interval between transplantation and immunization [474]. Vaccination during the first 6 months unfortunately was ineffective. Similarly, patients with GvHD were less likely to undergo seroconversion. Thus, early after transplant an agent such as a neuraminidase inhibitor may be more useful in protecting susceptible patients than immunization. It is currently advised that all stem cell transplant candidates and recipients receive a seasonal trivalent inactivated vaccine [134]. In addition, vaccination of family members and close or household contacts is recommended during each influenza season. This immunization schedule should start the year prior to the BMT and continue for at least 24 months after BMT, generally for as long as the patient is immunosuppressed [134]. If the immunization occurred during an influenza outbreak, chemoprophylaxis, if feasible, should be provided for 2 weeks after influenza vaccination while the immunologic response to the vaccine develops.
Recommendations for vaccination and chemoprophylaxis are identical for autologous and allogeneic transplant recipients.
Parainfluenza Viruses
Human parainfluenza viruses (PIVs) are enveloped, single-stranded RNA viruses, which are members of the Paramyxovirus family (along with RSV, measles, and mumps). Of the four major antigenic serotypes, three are common respiratory pathogens frequently causing upper (type 1 and 2) and lower (type 3) respiratory illness in the general population. Infections are seasonal, especially during the fall. The target tissues are the mucosal epithelium of the nasal and oropharyngeal passages, but infection can extend to bronchi and bronchioles in more severe cases. Neutralizing antibody and cytotoxic T-cell responses are immunoprotective. The virus can be isolated from infected secretions by cell culture techniques coupled with immunofluorescence.
Incidence and Outcomes
Parainfluenza virus is a frequent cause of respiratory illness in children less than 6 years of age. The disorder presents with a variety of symptoms, ranging from upper respiratory tract infections to croup and pneumonia [475]. By 30 months of age, nearly every child will encounter this virus, but immunity is incomplete and reinfection occurs throughout life. When reinfection occurs in later life, the infection course is generally less severe. Parainfluenza virus infections have been reported following BMT [475–478]. The reported incidence among BMT and cancer patients has ranged from 2 to 5 % of patients. The largest review of PIV in BMT patients was conducted over 16 years from 1974 to 1990 at the University of Minnesota. Over 1,200 patients were screened for common respiratory viruses, and PIV was isolated in 27 patients by either nasopharyngeal cultures or bronchoalveolar lavage. Approximately 30 % of patients had respiratory symptoms localized to the upper respiratory tract, characterized by fever, cough, coryza, wheezing, and shortness of breath. The remaining 70 % developed lower respiratory tract infections, of which 32 % developed respiratory failure. Diffuse pulmonary infiltrates on chest X-ray and sinus opacification, in addition to cough, fever, wheezing, and shortness of breath characterized lower respiratory tract infections. As with other CRVs, development of pneumonia is associated with a poor outcome, with the majority of patients dying. This is in contrast to infection localized to the upper respiratory tract, which all patients survived. Unlike other CRVs such as RSV, infection with PIV was not seasonal but occurred in nearly every month of the year. The most common subtype identified was parainfluenza type 3, which is consistent with other reports in the literature.
The M.D. Anderson group has also reviewed the frequency and outcomes of PIV infections in their cancer and BMT population. Parainfluenza was isolated in 3 % of 265 adult BMT patients during 1991. Again, PIV type 3 was identified in all patients. Progression to lower respiratory tract infections was associated with a 50 % mortality. More recently, this group published updated results spanning a 5-year period. Parainfluenza was isolated in 61 patients (5.2 % of the total screened population). Almost half of the patients evaluated with PIV developed pneumonia, with an associated mortality rate of 34 %.
Based on these studies, it is apparent that patients at greatest risk for PIV infection are those who have recently undergone BMT, that is, who are less than 100 days from BMT, and those undergoing allogeneic BMT. In addition, patients with pneumonia were more likely to be lymphopenic than patients with localized upper respiratory tract infections (78 versus 55 %, p < 0.005). Other risk factors evaluated but not found to be statistically predictive for PIV pneumonia were patient age, presence of GvHD, type of immunosuppression, and the presence of neutropenia [478].
Treatment
There is no known effective antiviral treatment, although ribavirin may have some activity [393–395, 425, 429]. Ribavirin has been used with mixed results in PIV infections. The Minneapolis group treated nine patients (33 % of the total PIV-infected population) with aerosolized ribavirin for a median of 10 days. The survival among those patients (78 %) was no different from that patients not treated with ribavirin. This study was uncontrolled and was not intended to test the value of ribavirin therapy. The Texas group also treated some patients with ribavirin, achieving success in 60 % of patients. At this juncture the role of ribavirin in PIV infections is unclear. Its role should preferably be in those patients with upper respiratory tract infections to attempt to prevent progression to a lower respiratory tract infection. However, in patients who have already developed a lower respiratory tract infection and are critically ill, it is also worth attempting. There are also limited data supporting intravenous ribavirin in the management of PIV infection [479].
Metapneumovirus
Human metapneumovirus is a single-stranded RNA paramyxovirus that causes respiratory tract infections in children, the elderly, and immunocompromised adults [480–483]. It has been identified as the cause of upper and lower respiratory tract infections worldwide during the late winter months (January to April). In one study of lung transplant recipients, infection was found in bronchoalveolar lavage fluid of 5 % of symptomatic patients [480].
Infection manifesting as acute pneumonia with diffuse alveolar damage and hyaline membrane formation has been described in lung and heart lung transplant recipients [484, 485]. In BMT recipients, one study isolated human metapneumovirus by RT-PCR in 26 % of symptomatic patients undergoing bronchoscopy and was associated with an 80 % mortality rate [486]. Infection usually occurred within the first few weeks after transplant and was characterized by fever, nasal congestion, cough and the rapid development of hypoxia, hypotension, and a rapidly worsening pneumonia. Pleural effusion and nodular infiltrates can be seen, which can help differentiate the infection from RSV. Prolonged asymptomatic infection after BMT has also been described [487]. Ribavirin has been demonstrated to decrease human metapneumovirus replication in the lungs in a mouse model [488]. Intravenous ribavirin has been effective in the treatment of metapneumovirus infection in lung transplant recipients [489].
Hepatitis Viruses
Hepatitis viruses are RNA (hepatitis A, C, and G) or DNA (hepatitis B) enteric pathogens, which can be transmitted via the fecal–oral route or sexual contact. Transmission through infected blood products is also problematic and represents a serious challenge for the management of patients with hematologic malignancies requiring blood product support. A summary of recommendations for the management of the various hepatitis viruses are listed in Table 53.6.
Table 53.6
Recommendations for screening, surveillance, and treatment of Hepatitis viruses
Virus | Screening | Surveillance | Prophylaxis | Treatment |
|---|---|---|---|---|
A | Recipient | N | N | IVIG, Hep A vaccine |
B | Donor/recipient | Y | Y | Lamuvidine, famciclovir |
C | Donor/recipient | Y | N | Pegylated interferon, ribavirin |
G | N | N | N | Unknown |
Hepatitis A
Hepatitis A is a member of the Picornaviridae family. This nonenveloped single-stranded RNA virus replicates in the intestinal tract and then is transported to the liver, which is the major target tissue. It can cause acute hepatitis characterized by jaundice or can present subclinically. In contrast to hepatitis B and C, only rare cases of hepatitis A transmission by blood products have been described, and therefore routine screening of blood products for hepatitis A virus does not occur. Transmission of the virus is thought to occur via the oral–fecal route. In addition, hepatitis A has a long incubation period of 15–50 days, with the virus excreted in the feces for 1–2 weeks prior to the onset of clinical illness and for at least 1 week afterwards [490], which emphasizes the need for good hygiene practices. The typical presentation of hepatitis A virus infections includes fever, malaise, nausea, vomiting, arthralgias, and anorexia [491]. In some patients flu-like symptoms may also be present. These symptoms tend to abate with the onset of jaundice. Rarely, acute liver failure, cholestatic hepatitis, or relapsing hepatitis can occur.
Management of acute hepatitis A infection is primarily symptom control. In otherwise healthy patients, resolution of the infection will occur in 3–6 months. The primary control measure is prevention. Patients traveling to hepatitis A endemic areas should receive either IVIG prophylaxis or hepatitis A vaccination. Similarly, immunocompromised patients who come into contact with hepatitis A-infected patients should consider prophylaxis. Passive immunization with IVIG has been the primary treatment of choice for over 50 years. Intravenous immunoglobulin is most effective when given within 2 weeks of exposure and is the fastest way to achieve hepatitis A antibodies. The alternative to IVIG is hepatitis A vaccine, of which there are two types used around the world, including the live attenuated vaccines and the inactivated vaccine. In the United States there are two commercially available inactivated vaccines, Havrix (Glaxo Smith Kline) and Vaqta (Merck). These vaccines produce very high levels of antihepatitis A virus, significantly higher than can be achieved with IVIG [491]. Following the first dose, antihepatitis A virus is detectable in the serum within 15 days in most patients. In the majority of patients, seroconversion is complete 1 month following injection, and persisting antibodies can be detected at 1 year. As with other vaccinations, immunosuppressed patients tend to be less responsive.
Indications for immunization include travel to endemic areas, chronic liver disease, and clotting factor disorders likely to require concentrate replacement. At this time, routine hepatitis A immunization is not recommended for BMT patients [438]. There are limited data on the safety of the vaccine in this population; however, consideration can be given to administration of the hepatitis A vaccine to patients older than 2 years who are at least 12 months post-BMT and who are at increased risk of for hepatitis A and its adverse consequences (e.g., persons with chronic liver disease, including chronic GvHD, and children living in areas with consistently elevated hepatitis A incidence) [490].
Testing for Hep A IgG in candidates and donors is generally not recommended as it has no impact on transplant outcomes. However, testing for Hep A IgM is generally indicated as part of the workup for acute hepatitis in HCT candidates because of the increased risk for sinusoidal obstruction syndrome following myeloablative conditioning regimens. Donors who test positive should have the transplant delayed because of a high risk of transmission and increased morbidity and mortality.
Hepatitis B
Hepatitis B, a member of the Hepnaviridae family, is a small DNA virus, which can cause acute self-limited primary infection and resolve or can become a persistent infection for years or even a lifetime. The virus can be transmitted by percutaneous inoculation via needles, blood transfusion, or contact of mucosal surfaces with blood and other body fluids, including sexual contact. Persistent infection may be accompanied by no hepatic dysfunction, but chronic hepatitis is common and can be associated with a risk for hepatocellular carcinoma. Persistent viremia can pose a risk for transmission by blood transfusion. With routine donor screening for hepatitis surface antigen (HbsAg) and hepatitis B core antibody (anti-HBc), the risk for hepatitis B from a blood transfusion has diminished tremendously. In addition, increasing immunization with hepatitis B vaccines in the community has led to a decrease in the number of patients infected with hepatitis B and hence a decrease in the number of contaminated transfusions. Less than 5 % of posttransfusion hepatitis today is caused by hepatitis B.
The overall incidence of hepatitis B has declined from a peak of 11.5 cases per 100,000 in 1985 to 1.5 cases per 100,000 in 2007, with an estimated 800,000 to 1.4 million chronically infected Americans [492, 493]. The prevalence of hepatitis B virus (HBV) is geography dependent. In the United States approximately 0.1–0.5 % of the population are hepatitis B carriers [493] compared ≥8 % in all of Africa, the Middle East (except Israel), South and Western Pacific islands, the interior Amazon River basin, and certain parts of the Caribbean (Haiti and the Dominican Republic) [494]. Based on these data, the chance of a patient undergoing chemotherapy also being a chronic hepatitis B carrier is increasing. Similarly, the likelihood of a BMT candidate having hepatitis B or being exposed to an HbsAg positive donor is increasing.
Chronic hepatitis B carriers who undergo chemotherapy or BMT have been shown to have an increased risk of liver-related morbidity and mortality [495–505]. At the completion of chemotherapy or BMT conditioning, the patient becomes immunosuppressed. At this time there is an increase in hepatitis B virus replication in the liver, leading to an increased viral load. On recovery postchemotherapy, there is immune reconstitution, which is associated with an acute hepatitis flare. Immune recovery causes an immune-mediated destruction of liver cells containing HBV. The severity of hepatocyte destruction is directly proportional to the viral load in the liver [493, 496, 499]. This results in an acute exacerbation of hepatitis and may progress to fulminant liver failure.
Studies have been performed in lymphoma patients with concomitant HBV undergoing standard chemotherapy. In this population, the development of impaired liver function (as assessed by increased liver function tests) occurred in more than 50 % of patients studied. Liver-related mortality was 5 %, with reactivation of HBV thought to be the cause of death [499]. Similarly, there are data characterizing an increased risk of HBV morbidity and mortality in HBsAg-positive BMT recipients [506–513].
In the solid tumor population, prediction of who will reactivate HBV is difficult. The most useful marker of reactivation is HBV viral DNA level [498]. Close monitoring of hepatitis markers is essential. Management strategies in the HBV-positive patient undergoing chemotherapy include as the first step removal of those agents likely to potentiate HBV reactivation. Corticosteroids are a common component of chemotherapy regimens and are known to stimulate viral reactivation [514]. If possible, steroid therapy should be avoided, but this may not always be possible. Continuous low-dose corticosteroids have been attempted and are generally no better than pulse steroids [499]. Similarly, daily or weekly schedules have not been shown to be better than schedules calling for administration every third to fourth week. Other drugs that increase the risk for reactivation/exacerbation of hepatitis B include rituximab [515] and alemtuzumab [516].
The alternative approach is to prophylactically administer antiviral agents to prevent HBV reactivation. Interferon has demonstrated efficacy in hepatitis B carriers (but is less effective in Asian hepatitis B carriers). One of the important side effects of interferon is myelosuppression, and for this reason it is often contraindicated in the already myelosuppressed cancer patient. Alternatives include the nucleoside analogues lamivudine and famciclovir. Both of these agents interfere with viral DNA replication, leading to inhibition of serum HBV viral DNA levels, with consequent improvement in liver function tests (LFTs) [517–523].
The most effective treatment strategy is targeting those patients at greatest risk (identified by screening patients for past and current infection with HBV) and administering prophylaxis to prevent viral reactivation [524]. Patients with evidence of disease at the time of chemotherapy, as demonstrated by viral replication (HBV-DNA-positive) should receive an antiviral agent such as lamivudine (100 mg orally daily) or famciclovir (500 mg three times daily). Unfortunately, on many occasions the viral load in the liver is too great to be affected by this treatment. Carriers of HBV can either be treated prophylactically with the nucleoside analogues or closely monitored postchemotherapy for evidence of viral replication. In those patients receiving prophylactic therapy with the nucleoside analogues, therapy should be continued for at least 6 months following completion of chemotherapy [524].
Management of patients undergoing autologous BMT is similar to conventional chemotherapy. Delayed immune reconstitution may lead to a greater risk of reactivation, but the risk is not as great as that seen in allogeneic BMT recipients. Allogeneic BMT in the HBsAg-positive patient is somewhat more complicated. At the time of transplantation, the patient may be actively infected with hepatitis B virus (HBsAg-positive) or alternatively latent HBV may be reactivated following transplantation (HBsAg-negative) [524]. In the allogeneic setting, the hepatitis B status of the donor must also be considered; otherwise, liver failure secondary to hepatitis B infection is a possible consequence.
The mechanism of viral reactivation is very similar to that seen after chemotherapy. BMT-conditioning chemotherapy causes ablation of both the bone marrow and the immune system, leading to increased viral replication. On recovery of the immune system, an acute flare may occur. Similar flares have been documented when tapering cyclosporin A immunosuppression [495]. In a study of 43 hepatitis B virus carriers undergoing autologous or allogeneic BMT with similar preconditioning regimens, none of the autologous BMT patients developed fulminant hepatitis, compared to five allogeneic patients. Impaired LFTs correlated with cyclosporin A withdrawal [495] and could not be controlled by cyclosporin A dose escalation. Potential signs of a hepatitis flare include increases in LFTs, particularly serum aspartate aminotransferase and alanine aminotransferase (ALT). Hepatitis serology should also be monitored. Ominous signs for the development of fulminant hepatitis include changes in serology from HbeAgnegative to HbeAg-positive, the presence of HbcAb IgM, or detection of HBV-DNA [495].
Approaches to the prevention and treatment of HBV in allogeneic BMT patients are complicated and depend on both the donor and recipient hepatitis profiles. Prior to BMT, all patients and donors should have a hepatitis workup, including HBsAg and HBcAb. If the HBsAg is positive, HBeAb, anti-HBe, and HBV DNA (including quantification) should be assessed. Recommendations for management depending on the results of hepatitis screening have been reviewed [134, 525]. The key to management of BMT patients is comprehensive upfront screening and prophylaxis with a nucleoside analogue if infected.
In addition to lamivudine and famciclovir, newer agents, including adefovir dipivoxil, entecavir, telbivudine, and tenofovir, are effective in suppressing viral reactivation [526, 527]. Unfortunately, the emergence of drug-resistant variant viruses with mutations in the HBV polymerase gene has occurred with their use [528–530] and it is unclear whether any of these newer agents should be used for first-line preemptive therapy. An inactivated hepatitis B vaccine is highly protective. In nonimmunized exposed individuals, hyperimmune globulin can afford protection. Interferon can suppress hepatitis B replication although the magnitude of benefit is not great and is frequently temporary [531]. In one study an attempt to immunize BMT and leukemia patients against hepatitis B was unsuccessful [532]. In one case persistent hepatitis B was ablated in a patient who received a marrow transplant from a hepatitis B-immune donor [533]. Similar experimental data in mice have also been supportive of the concept that donor immunity can be adoptively transferred to BMT recipients [534].
In conclusion, when managing BMT recipients, consideration should be given to HBV infection in the differential diagnosis in post-BMT patients with abnormal hepatic transaminases not explained by the typical causes of hepatitis, for example, medications, GVHD, or other viral infections (CMV, HSV, VZV). Increases in hepatic transaminases are most likely at the time of immunosuppression tapering. Provided treatment and prophylaxis recommendations and guidelines are adhered to, the risk of developing HBV infection is low.
Hepatitis C
Hepatitis C is a lipid-enveloped single-stranded RNA virus related to members of the Flaviviridae family. Hepatitis C virus (HCV) can be transmitted via blood transfusions, intravenous drug abuse, and needlestick injuries, as well as by vertical transmission. The risk of transmission of HCV by a blood transfusion has decreased significantly following the introduction of screening processes in 1991. The magnitude of the decrease is such that the risk of transmission in donated blood that has passed all screening tests is now 1 in 103,000 [535] compared to 0.19–0.45 % prior to 1991 [536]. The seroprevalence of antibody to HCV in most developed countries is approximately 1–2 %. As with hepatitis B, there is significant geographical variation in seroprevalence rates, with higher rates of infection in Eastern Europe and Africa than in the United States. The Centers for Disease Control estimates that nearly 4 million people are infected with HCV in the United States [537]. There are six genotypes of HCV, each having a different geographical predominance. The most common genotypes in the United States are 1a and 1b; genotype 1b is also common in Europe. In Asia genotype 6 is most common, in South Africa genotype 5 is most common, in the Middle East and North Africa genotype 4 is most common, and among intravenous drug users the prevalence of genotype 3 is rising. Differences have been seen in the various genotypes in terms of responses to therapy, but there is currently no clear correlation of disease outcome and severity with genotype [538].
Hepatitis C can cause an acute, self-limiting, primary infection and resolve, or it can become a chronic infection. Approximately 85 % of HCV-positive patients develop chronic HCV infection (as documented by persistence of HCV RNA in serum) [538]. There are data demonstrating that cirrhosis develops in more than 20 % of chronically infected adults an average of 20 years after diagnosis of the acute infection [539]. Like hepatitis B, hepatitis C has been linked to hepatocellular carcinoma. Hepatocellular carcinoma occurs most commonly in those individuals who develop cirrhosis. Other complications seen commonly in HCV-infected people include elevated transaminases; symptoms of terminal liver failure, that is, portal hypertension and hepatic encephalopathy; and hepatic fibrosis. Hepatitis C has also been reported to cause extrahepatic manifestations, for example, porphyria cutanea tarda, mixed cryoglobulinemia, and glomerulonephritis [540].
Hepatitis C virus infection can be documented serologically by the detection of antibody directed against HCV in serum (anti-HCV) using enzyme immunoassay (EIA) methodology. These tests have been shown to detect anti-HCV in more than 97 % of infected individuals. The test is useful for demonstrating past or present infection but is not able to differentiate among acute, chronic, and resolved infection [541]. The EIA technique occasionally produces false positive results, and therefore confirmatory testing with a supplemental assay is often performed. In cancer patients and the immunocompromised, it is prudent to test for HCV RNA, as this population may produce a false negative result due to inability to produce sufficient antibodies for detection by EIA. The virus itself can be detected in blood through the use of reverse transcriptase polymerase chain reaction (RT-PCR) amplification of HCV RNA. This test enables detection of circulating HCV RNA early in the course of HCV infection, that is, 1–2 weeks following exposure. The presence of HCV RNA indicates an active infection. The final test is a quantitative assessment of the concentration of HCV RNA in the blood (viral load assessment). This test is only useful for determining response to therapy and does not correlate with the severity of hepatitis or overall prognosis.
Hepatitis C infection is relatively common in the cancer (prevalence up to 70 %) [523, 542–551] and BMT (3–70 %) [552–556] population, particularly those who were treated prior to 1991. These patients have been at greater risk of developing HCV infections than the general population because of the large number of blood products they received during or following therapy. Since the introduction of routine screening tests, the proportion of infected patients has decreased. Because of the large number of people infected with hepatitis C worldwide, the chance of a BMT patient or donor being hepatitis C positive is relatively high. Therefore, appropriate diagnostic and subsequent precautionary measures should be taken to avoid fulminant liver failure. Patients thought to be at risk for hepatitis C (i.e., those with increased ALT pretransplantation that is not explained by other likely diagnoses, those with a history of intravenous drug abuse, with a history of blood transfusion prior to 1991, or with multiple sexual partners) should be evaluated for hepatitis C infection by HCV RNA PCR detection technology prior to transplantation.
Patients who are infected with HCV and undergo BMT are at increased risk for the development of veno-occlusive disease (VOD) [556, 557]. The Fred Hutchinson Cancer Research Center (FHCRC) group published 10-year follow-up data on a cohort of 355 patients undergoing BMT in 1987–1988 to assess the impact of pretransplant HCV status on liver dysfunction, including VOD. Before transplantation 17 % of patients were HCV RNA-positive which increased to 32 % by day 100 following transplantation. Veno-occlusive disease occurred more frequently in those HCV RNA-positive pretransplant patients (87 %), compared to 66 % in those without HCV pretransplant (p < 0.004). Severe VOD was also increased in patients who were HCV positive prior to transplant compared with HCV-negative patients (48 versus 14 %, p < 0.0001) [556]. Many factors are known to contribute to the risk for developing VOD, and a multivariate analysis of these factors was conducted in this cohort of patients. The greatest risk factor in these patients was pretransplantation HCV infection, in conjunction with elevated serum aspartate transaminase (AST) levels (relative risk 9.6, p = 0.0001). Patients infected with HCV prior to transplantation who had normal AST levels had no greater incidence of VOD. European investigators have not found the same association, and it is postulated by the FHCRC group that this may be due to a lower dose intensity of chemotherapy in the European studies [552, 558–560].
HCV infection may also have a significant role in long-term survival after HCT. The first study which directly assessed the impact of HCV infection in long-term survivors after HCT was conducted by Ljungman et al. [561]. The diagnosis was based on the HCV positivity either by PCR for HCV RNA or by second-generation ELISA and RIBA or EIA supplemental assay. Of 161 surviving patients transplanted between 1978 and 1991, 28 (17.4 %) were found to have chronic HCV infection. No signs of severe progressive liver disease were shown among the patients included in this study with a follow-up median time of 6.1 years [561]. Thomas et al. with an average follow-up of 6 years found no evidence of cirrhosis [562]. In 1999, the Seattle group reported a cohort of 355 patients that underwent HCT between 1987 and 1988 from which 113 (32 %) were HCV RNA-positive by day 100 posttransplant. During 10 years of follow-up, no patients developed clinical evidence of liver disease, and HCV infection did not impact the actuarial survival of long-term survivors over this time period [563]. It was thus concluded that HCV infection was not associated with excess mortality over 10 years of follow-up. However, the same group observed the development of cirrhosis leading to hepatic decompensation and hepatocellular carcinoma in HCV-infected, marrow transplant recipients, surviving beyond 10 years. Among 3,721 patients who survived 1 or more years after HCT, 31 developed cirrhosis. Cirrhosis was attributed to HCV in 15 of 16 patients presenting more than 10 years after HCT [564]. HCV infection ranked third, behind infection and GVHD, as a cause of late death [565]. Moreover, cirrhosis was diagnosed beyond 10 years after transplantation in 13 of 15 patients and 3 cases of hepatocellular carcinoma was observed in this cohort. Ivantes et al. also found cirrhosis on a smaller group of patients followed up more than 10 years after HCT [566]. Thus, HCT recipients with HCV infection present a higher risk of earlier cirrhosis.
It is recommended that all candidates for HCT with HCV infection must be assessed for evidence of chronic liver disease [134]. Liver biopsy is advised in the setting of associated iron overload, history of excessive alcohol intake, history of hepatitis C > 10 years, and clinical evidence of chronic liver disease. Patients with evidence of cirrhosis should avoid conventional Cy or TBI containing conditioning regimens as they are associated with a 9.6-fold increase in fatal sinusoidal obstruction syndrome [556]. However, even with reduced intensity conditioning regimens, there is a significant mortality risk for patients with cirrhosis [567].
There are currently no good treatment options for HCV-infected BMT recipients. Standard HCV treatment for the general population includes interferon-α, either alone [568–574] or in combination with ribavirin [575–577]. Interferon (3 million units subcutaneously three times per week for 12 months) is active and has been shown to reduce the level of HCV viremia and improve hepatic transaminase abnormalities [568–570]. Long-term remissions can occasionally be seen (in 15–25 % of cases) [541], although unfortunately more than 50 % of patients who respond initially relapse after treatment cessation. Predictors of a poor response to interferon include a high HCV RNA titer and HCV genotype 1 (the most common genotype in the United States). The combination of ribavirin (1,000 mg/day divided into two doses for patients weighing less than 75 kg; 1,200 mg/day divided into two doses for those over 75 kg) and interferon has produced virological response rates that were significantly higher with double therapy than with monotherapy (36 versus 18 %, p < 0.047) [576], and the responses were sustained over 12 months. In most clinical situations, combination therapy is preferred. Currently, the favored treatment option is the combination of pegylated alpha interferon and ribavirin which produced a sustained virological response rate of approximately 55 % [578, 579]. Responses were better in genotype 2 or 3 infections versus genotype 1 infections (approximately 80 % response with a 24-week treatment duration versus 50 % with a 48-week duration, respectively).
These treatment options are often not possible in allogeneic transplant candidates. Interferon therapy is typically contraindicated in allogeneic BMT patients within 6 months of transplantation because of an associated increased risk of GvHD and mortality [580–582]. In addition, ribavirin monotherapy has been shown to be ineffective in the general population [583–596], although there is one small case series that demonstrates efficacy in BMT patients [587]. If treatment is not possible with either of these agents, this should not necessarily be considered a contraindication to transplantation. In general, the course of chronic liver disease is insidious, and progress is slow for at least 10–20 years. The determination of whether transplantation should proceed and of when to transplant is dependent on the degree of underlying liver dysfunction, which should be established prior to transplantation. Workup of liver dysfunction should include laboratory estimations, namely LFTs and coagulation assessments, as well as physical examination and if indicated, a liver biopsy. A liver biopsy is indicated in those patients in whom there is a clinical suspicion of cirrhosis, in patients who are likely to have had HCV infection for longer than 15 years (long-term cancer survivors in the era where there was no screening for hepatitis C) and in those known HCV-infected patients with a history of excessive alcohol intake [524]. If a diagnosis of hepatic fibrosis or cirrhosis is made, the conditioning regimen and type of transplantation procedure performed need to be reassessed. With the risk of severe VOD of the liver and the potential for multiorgan failure and subsequent death with standard myeloablative conditioning regimens, a nonmyeloablative transplant should be considered in this setting.
In the allogeneic BMT setting, the HCV status of the donor must also be evaluated. Hepatitis C virus transmission to recipients of HCV RNA-positive allogeneic and syngeneic marrow products [588] occurs universally. Within days of marrow reinfusion, patients develop viremia, although evidence of clinical and biochemical hepatitis may take weeks to months to develop. Damage to hepatocytes in HCV infection is mediated predominantly by HCV-specific cytotoxic T cells, and therefore biochemical abnormalities will only become apparent on engraftment and reconstitution of cell-mediated immunity [556, 557, 589]. If time permits, the best action is to treat the donor with interferon with or without ribavirin in an attempt to render the donor’s serum HCV RNA undetectable at transplantation. However, treatment of HCV infection may take 6–12 months, time which is not always available in transplantation because of the underlying nature of the malignant disease. Again, donor HCV seropositivity is not a contraindication to transplantation, priority being allocated to an HLA match, whether donor and recipient are HCV-positive or -negative.
One of the issues that has yet to be addressed in a large study is how to manage the long-term survivor of cancer or bone marrow transplantation. As supportive care technologies improve, the length of overall survival is increasing, which will lead to a population of patients with the potential to develop cirrhosis. As mentioned, blood transfusion products were not screened for HCV prior to 1991. With the insidious nature of the disease, the implications of cancer treatment and transplantation in this group may not have been truly realized before mid-2000. Common sense tells us that those patients developing signs of cirrhosis and liver failure should be treated in the same manner as the general HCV-infected population. In allogeneic BMT patients, this will be determined by degree of immunosuppression and the presence of GvHD. In addition, all long-term survivors of cancer with chronic viral hepatitis should be offered vaccination because of the increased risk of fulminant hepatitis should they develop acute hepatitis A [590, 591].
Hepatitis G
Hepatitis G virus (HGV) is a RNA virus that belongs to the Flaviviridae family [592, 593] and is closely related to the GB virus-C (GBV-C) [594]. It prevails worldwide, and the viral RNA has been detected in 1–5 % of volunteer blood donors [594, 596]. The prevalence of HGV in patients undergoing BMT ranges from 18 to 61 % [597–602]. Following BMT, immunosuppressed patients have been reported to have an increased risk of HGV infection after transfusion-related exposure [598, 603]. This has not translated into accelerated HGV-RNA replication [602].
Currently, the clinical relevance of HGV infection and its long-term effect on BMT patients remain unresolved. HGV has not been shown to affect engraftment kinetics, which indicates no direct effect of HGV on hematopoietic progenitors [604]. The data on the effect of HGV on liver function are conflicting. There are data demonstrating no effect of HGV on the clinical and biological diagnosis of hepatitis in immunosuppressed patients in several studies [601, 603]. However, there are also data in pediatric patients demonstrating increased ALT levels in the absence of other known causes [597]. The maximum ALT levels were statistically higher than those of uninfected patients, although the mean values of ALT showed no difference. This suggests that the effect of HGV on ALT is transient and self-limiting [597].
At present blood for transfusion is not routinely screened for HGV, which is not considered to be a serious pathogen nor a significant cause of hepatitis. Clearly, additional large studies of HGV-infected immunocompromised patients are needed to definitively characterize the effect of HGV on engraftment and hepatotoxicity.
Other Viruses
Papovaviruses (Polyomaviruses)
Infection by JC and BK viruses occur early in childhood and are widespread. In most individuals it is asymptomatic, but the viruses persist in the kidney and urogenital epithelium. Asymptomatic virus excretion increases in immunocompromised patients [605, 606]. The JC virus has been associated with progressive multifocal leukoencephalopathy and has been described in patients with chronic lymphocytic leukemia or lymphoma and following BMT [607–609]. Dissemination with involvement of mutiple visceral organs, including the lungs, has also been noted [605]. The BK virus has been associated with hemorrhagic cystitis, especially in allogeneic BMT patients, in whom the risk is three times as high as in similarly treated autologous BMT recipients [610–614]. Occasionally adenovirus type 11 can also be associated with hemorrhagic cystitis [611]. Both humoral and lymphoproliferative responses to active viral shedding have been noted, but whether these are immunoprotective remains uncertain [615]. At present, there is no known effective therapy, although cases of response to vidarabine, ribavirin, ciprofloxin, or cidofovir have been reported.
Parvoviruses
Human parvovirus B19 is a small DNA virus, which causes erythema infections during childhood. It is an occasional cause of arthritis and in patients with chronic hemolytic anemia can be a cause of a transient aplastic crisis. It is a potential cause of pancytopenia, rash, or chronic anemia in immunocompromised patients, including leukemia and BMT patients [616–624].
Intranuclear lesions within erythroid precursors in bone marrow specimens have been noted in acutely infected patients [625].
Marrow recipients with GVHD may have chronic infection. The infection can be documented by detection of virus-specific antibody against the B19 antigen by ELISA or of viral DNA by PCR. The virus has been detected in respiratory secretions, bone marrow, and blood. There is no specific antiviral treatment, but reducing the intensity of chemotherapy can be helpful [620], and immune globulin may also be of benefit.
Retroviruses
Human immunodeficiency virus is a retrovirus that is the causative agent of AIDS. Sexual transmission and transmission via blood transfusion or organ transplantation has been well documented. During the early 1980s, prior to the development of a screening serologic test for blood products, patients with hematologic malignancies receiving blood product support were susceptible for contracting HIV infection. Subsequent to 1985 with routine screening, the risk of transmission via a blood product is 1 in 500,000 or lower. Several cases of AIDS in patients with hematologic malignancies have been documented [626]. It is uncertain that the course of illness in immunocompromised patients is substantially different than in nonimmunocompromised patients. The association of HIV infection with Kaposi’s sarcoma and aggressive non-Hodgkin’s lymphoma has been noted, especially among those with low CD4 counts for more than 1 year [627–630]. There has been limited experience performing bone marrow transplantation in patients with HIV-associated malignancies [631–633]. In some patients, cytoreductive therapy to ablate all lymphohematopoietic-derived cells (the reservoir of latently infected cells) combined with antiviral therapy has had a marked antiviral effect, whereas in others in whom the virus is not very susceptible, the effect has been marginal. Most of the beneficial effects have been transient, and generally patients have succumbed from transplant-related complications or opportunistic infection. With the development of better antiviral agents and the engineering of donor marrow stem cells with genes that confer resistance to viral infection, this might be more promising in future investigations.
In the era of highly active antiretroviral therapy (HAART), there has been a drop in the rates of opportunistic infection and Kaposi’s sarcoma. Of interest, however, is that there has not been a concomitant fall in the incidence of AIDS-related lymphomas, at least so far [634–637], with the exception of a decline in central nervous system lymphomas [627, 628]. Unfortunately, most cases present at an advanced stage and have a very poor prognosis, which also has not changed in the era of HAART [634, 638]. In a subset of patients with CD4 counts above 100/ml, higher response rates and longer survival have been noted [639]. Since HAART therapy may preserve immune function for long intervals, diagnosis of lymphomas at an earlier stage may in the future lead to improved treatment outcomes.
Currently, the presence of HIV in patients with a malignancy treated by transplantation does not automatically exclude the patient from this potentially life-saving procedure. In patients with controlled HIV disease, autologous transplantation is a reasonable, with transplant related mortality similar to non-HIV infected patients [640–644]. The data for allogeneic transplant, however, is limited although it is feasible [645]. Management of these patients should be coordinated with a HIV specialist and preferably on a clinical trial.
Human T-cell lymphotropic viruses (HTLV)-1 and -2 are type C oncoviruses present in widely scattered populations, including those of the islands of southwestern Japan and the Caribbean basin, where infection is endemic. Screening of blood donors in the United States indicates a small but detectable seroprevalence. Transmission is by blood transfusion, sexual contact, and in utero. Although most infections are asymptomatic, HTLV-1 is the etiologic agent of adult T-cell leukemia/lymphoma [646–648]. There have also been associations between HTLV-2 and malignancy [648], but its role in pathogenesis is uncertain. Adult T-cell leukemia/lymphoma occurs in only a small minority of HTLV-1 carriers. The lifetime risk is estimated to be between 2 and 5 % [646].
West Nile Virus
West Nile virus is a flavivirus first detected in the northeastern United States in 1999 and since then has caused over 2.5 million cases of infection in outbreaks of infection in the late summer and early fall throughout the Unites States [649, 650]. In humans, most infections are asymptomatic. However, 20 % of infected patients develop West Nile fever, manifested by malaise, anorexia, nausea, myalgias, headache, and occasionally lymphadenopathy [651]. One in 150 symptomatic patients develop meningitis or encephalitis, presenting as photophobia, phonophobia, and meningismus [652]. Patients with encephalitis develop altered mental status, cranial nerve palsies, seizures, and movement disorders. A minority of patients abruptly develop asymmetric weakness that may progress to flaccid paralysis associated with hyporeflexia or areflexia, mimicking poliomyelitis [650, 653, 654]. Acute neuromuscular respiratory failure may develop, with a mortality rate of more than 50 % [652]. Hemorrhagic fever has been described as well, similar to syndromes seen with other flaviviruses [655].
Diagnosis of West Nile virus infection in immunocompetent hosts may be made serologically or by reverse transcriptase PCR. An IgM antibody capture assay is available and becomes positive in CSF 3–5 days after onset of symptoms in nonimmunosuppressed hosts [656, 657], before serum antibody develops; CSF IgG is seen approximately 5 days later. Antibody presence may be confirmed with viral neutralization studies. IgM antibodies may persist in serum for up to 12 months after infection resolution, and IgG may persist for years. Immunocompromised patients demonstrate delayed seroconversion, making diagnosis of acute infection difficult at times. Nucleic acid testing in plasma or CSF is the most useful diagnostic test in this setting [658].
Several cases of West Nile virus infection have been reported in recipients of BMT [656, 659]. In most well-described cases, infection occurred 3–5 months after engraftment while on calcineurin-based prophylaxis against or treatment for chronic GVHD. Fever, lethargy, and progressive bilateral extremity weakness were noted. Hyporeflexia or areflexia were present, and CSF contained 0–6 white blood cells/μL. Serologic studies were negative in CSF and blood in most cases (including IgM assays). Diagnosis of West Nile virus infection was made by PCR performed on serum and CSF. All of the described patients died.
There are no antiviral agents that have proven efficacy in the treatment of West Nile virus infection. Ribavirin possesses in vitro activity but demonstrates poor clinical efficacy [660, 661]. Intravenous immunoglobulin with high titers of anti-West Nile virus antibodies were demonstrated to have significant clinical benefit in animal models. However, immunoglobulins derived from US donors has been ineffective in treating acute infection [649, 662, 663]. Overall mortality from West Nile virus are 4–20 % with significantly higher rates in immunosuppressed patients [652, 664].
Summary
The clinical significance of viral infections in patients with hematological malignancies has assumed greater importance during the last 2 decades. In part, this is related to more frequent recognition of viral syndromes due to improved techniques of documenting various viral pathogens by more accurate and more accessible diagnostic assays. It is also attributable to increasing use of dose-intensive or immunosuppressive treatment regimens, which have greater attendant immunosuppression and susceptibility for viral infection. With increasing awareness of the substantial morbidity due to these viral pathogens, it has become extremely important to recognize these infections. With the expanding array of antiviral agents, vaccines, and immune modulators, it has now become possible to intervene therapeutically to prevent infection, reduce morbidity, or prevent death from these pathogens. With continuing investigations it is likely that a number of unexplained syndromes encountered in patients with hematologic malignancies will be identified in the future as having a viral etiology. These advances hopefully will further improve our supportive care in this patient population and permit further strides in the control of hematologic malignancy.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


