Vascular endothelial growth factor (VEGF) is, to date, the key element in the pathogenesis of renal cell carcinoma (RCC). VEGF pathway activation is responsible for the recruitment, migration, and expansion of endothelial cells, with this angiogenesis tumor model being characteristic of RCC. Different strategies have been developed for almost a decade to block the VEGF pathway in this setting. Four different compounds were approved for metastatic RCC (mRCC) in the past 6 years: bevacizumab, sunitinib, sorafenib, and pazopanib. Axitinib and tivozanib are also promising compounds under evaluation. The revolution in the management and prognosis of patients with mRCC is ongoing.
Improved understanding of the biology and pathogenesis of renal cell carcinoma (RCC) has confirmed the role of vascular endothelial growth factor (VEGF) and its related pathway elements in the angiogenesis that promotes RCC pathogenesis. RCC tumor proliferation and survival have been shown to be largely mediated through this pathway. Therefore, inhibition of the VEGF signaling pathway is a therapeutic target for patients with RCC.
Targeted agents directed toward VEGF/VEGF receptor (VEGFR) pathways have become the mainstay of systemic treatment for metastatic RCC (mRCC), having largely replaced cytokine therapy because of improvements in both efficacy and tolerability. VEGF pathway inhibition relies either on VEGF ligand-binding blockade or on inhibition of its receptor, VEGFR.
Several questions are currently being assessed in this setting: which VEGF/VEGFR inhibitor is more potent and well tolerated? can this combination be sustained? what are the future promising agents for VEGF-targeting strategies to combat RCC? and lastly what is the best sequence of VEGF inhibitors to use?
This article focuses on the development, underlying rationale, and clinical data for the FDA-approved VEGF ligand-binding antibody (bevacizumab), small-molecule tyrosine kinase inhibitors (sunitinib, sorafenib, and pazopanib), and other drugs under development (axitinib and tivozanib) that block the intracellular domain of the VEGFR in the management of patients with RCC.
VEGF biology
Von Hippel-Lindau and VEGF
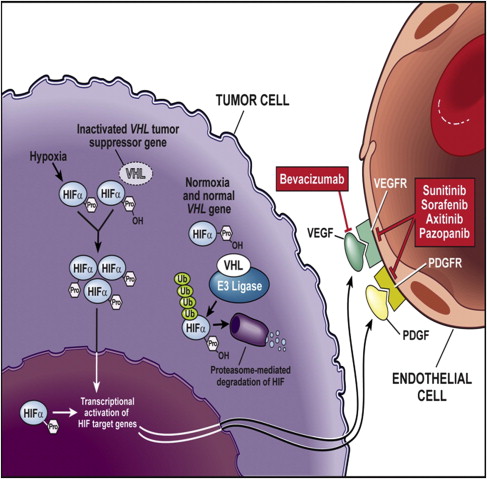
First isolated in the hereditary syndrome Von Hippel-Lindau (VHL) disease, the importance of the VHL tumor-suppressor gene emerged in sporadic clear cell RCC. VHL gene inactivation has been observed in 84% to 98% of sporadic RCCs. These observations are specific to clear cell RCC histology; VHL mutations have not been observed in other subtypes of RCC. Biallellic VHL gene inactivation has, therefore, been considered a key event in clear cell RCC oncogenesis according to the two-hits carcinogenesis Knudson model. The VHL protein regulates normal cellular responses to hypoxia via hypoxia-inducible factor α (HIF-α). When oxygen levels are normal, oxygen content in the blood regulates the formation of VHL protein complexes, which target HIF-α for degradation by proteasomes. Therefore, proangiogenic factors are not released. However, mutation or inactivation of the VHL protein disrupts the ability to degrade HIF-α in the presence of normal oxygen levels, leading to excess accumulation of HIF-α, and resulting in the overproduction of proangiogenic factors, such as VEGF. Therefore, inactivation of VHL function activates the hypoxia-response pathway. This pathway corresponds to transcriptional activation of a variety of genes involved in tumor proliferation, including VEGF ( Fig. 1 ). VEGF is a key player in promoting tumor-associated angiogenesis.
VEGF Function
VEGF is a growth factor that exerts its biologic effects primarily on vascular endothelial cells. It is part of the VEGF family of ligands, including VEGF-B, VEGF-C, and VEGF-D, which bind to one or more of the various VEGFRs (see Fig. 1 ). On ligation to its receptor, VEGFR-2 can induce growth, proliferation, and migration of endothelial cells, and promote the survival of immature endothelial cells via inhibition of apoptosis. It also increases vascular permeability. As a key proangiogenic molecule, VEGF plays an important role in several physiologic processes, such as embryogenesis, skeletal growth, and wound healing, and is the key mediator of angiogenesis in cancer.
Taken together, these scientific findings have led to the development of therapeutic inhibitors of VEGF in RCC. VEGF inhibition strategies rely either on VEGF blockade or on inhibition either of the VEGFR or of signaling of the downstream VEGFR. These approaches and their therapeutic results are described (see Fig. 1 ).
VEGF binding agents
Bevacizumab
Bevacizumab (Avastin, Genentech, South San Francisco, CA, USA) is a recombinant monoclonal antibody (mAb) IgG1 antibody that has been developed for humans from murine anti-VEGF mAb A4.6.1. The murine mAb A4.6.1 is specific for human VEGF, binding to all of the known isoforms of the ligand (eg, VEGF121, VEGF165, VEGF181, VEGF206). It is formed through alternative gene splicing, preventing it from binding to VEGFRs on vascular endothelial cells. In 1997, murine anti-VEGF mAb A4.6.1 was adapted for human use by site-directed mutagenesis, resulting in the production of bevacizumab. Bevacizumab is 93% human and 7% murine, and recognizes all of the major isoforms of human VEGF with a binding affinity of Kd = 8 × 10 −10 M (a similar affinity to the murine antibody). The binding ability of bevacizumab for VEGF is restricted to human, nonhuman primate, and rabbit VEGF. Sustained inhibition of VEGF with bevacizumab results in the regression of existing tumor microvasculature and normalization of surviving tumor vasculature, and inhibits the formation of new vasculature. It may also revert tumor-associated immune suppression and improve concomitant drug delivery into the tumor.
Bevacizumab has a terminal half-life of 17 to 21 days, with no dose-limiting toxicity when used as a single agent. The low interpatient variability and the modest effects of covariates on the clearance and volume distribution of bevacizumab support the current strategy of dosing bevacizumab based on body weight (mg/kg).
Phase II trials
Two key phase II trials have been conducted on bevacizumab use in treating RCC: AVF0890s and the RACE trial. The first trial, AVF0890s, was a randomized, placebo-controlled, double-blind trial of bevacizumab monotherapy conducted in patients with metastatic, predominantly clear cell RCC who were not optimal candidates for an interleukin (IL)-2 therapy or had not previously experienced response to this therapy. Between October 1998 and September 2001, 116 patients were randomized to one of three treatment arms: placebo (n = 40) or bevacizumab at either 3 mg/kg (n = 37) or 10 mg/kg (n = 39). This trial showed that the median time to progression was significantly longer for the 10-mg/kg bevacizumab arm than for the placebo arm (4.8 vs 2.5 months; hazard ratio [HR], 2.55; P <.001). The median time to progression for the 3-mg/kg bevacizumab arm was 3 months, and was not significantly greater than the placebo arm (HR, 1.26; P = .053). Four (10%) patients in the 10-mg/kg bevacizumab arm experienced partial responses of variable duration (6, 9, 15, and >39 months, respectively). This trial provided the rationale for the using 10 mg/kg of bevacizumab in treating RCC.
The phase II RACE trial evaluated bevacizumab in combination with erlotinib in patients with mRCC based on the rationale that VEGF has been implicated to have anti-epidermal growth factor receptor resistance. A randomized, double-blind, placebo-controlled trial was conducted at 21 sites in the United States. Eligible patients were enrolled from March 2004 through October 2004 to receive bevacizumab, 10 mg/kg, every 2 weeks, plus either erlotinib, 150 mg orally daily, or a placebo daily. The median progression-free survival (PFS) was not significantly improved by the addition of erlotinib to bevacizumab (8.5 months with bevacizumab plus placebo vs 9.9 months with bevacizumab plus erlotinib; HR, 0.86; 95% CI, 0.50–1.49). Furthermore, the addition of erlotinib to bevacizumab resulted in a similar overall response rate, which was 13% with bevacizumab plus placebo versus 14% with bevacizumab plus erlotinib. The addition of erlotinib to bevacizumab did not result in an improved duration of objective response (6.7 vs 9.1 months) or time to symptom progression (HR, 1.172; P = .5076). This efficacy has been the basis for testing bevacizumab in phase III trials.
Phase III trials
AVOREN trial
AVOREN was the pivotal phase III trial to evaluate the efficacy and safety of adding bevacizumab to interferon for treating mRCC. Bevacizumab was tested in combination with interferon to determine whether it would add efficacy to one of the standard treatments at the time of the trial’s design. Between June 2004 and October 2005, the trial enrolled 649 patients from 18 countries. Eligible patients had mRCC with a predominantly clear cell histology (>50% clear cells if mixed), underwent prior nephrectomy for primary RCC, a measurable or nonmeasurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST), a Karnofsky performance status of at least 70%, and no proteinuria at baseline (<0.5 g of protein in a 24-hour urine collection). Patients were randomized on a 1:1 basis to receive interferon (9 million international units) three times weekly plus a placebo, or bevacizumab at 10 mg/kg every 2 weeks plus interferon. The final analysis of PFS, which was performed at the scheduled time point for overall survival, showed that it was significantly improved by the addition of bevacizumab to interferon, for 5.4 to 10.2 months (HR, 0.63; P = .0001). This finding represents an 89% improvement in median PFS with bevacizumab plus interferon. The addition of bevacizumab to interferon also improves the overall response rate compared with interferon plus a placebo (31% vs 13%). Analyses of patient subgroups suggested that the addition of bevacizumab to interferon improves PFS in all subgroups analyzed. Improvements in PFS were observed in both favorable (n = 180) and intermediate (n = 363) Memorial Sloan-Kettering Cancer Center (MSKCC) risk categories (median PFS, 12.9 vs 7.6 months; HR, 0.60; P = .0039, and median PFS, 10.2 vs 4.5 months; HR, 0.55; P <.0001, respectively). A significant improvement was not seen in patients in the poor MSKCC risk category (n = 54; median PFS, 2.2 months with bevacizumab plus interferon vs 2.1 months with interferon plus placebo; HR, 0.81; P = .457). An improvement in PFS was observed in patients receiving bevacizumab plus interferon who either had a clear cell RCC histology (n = 564; median PFS, 10.2 vs 5.5 months; HR, 0.64; 95% CI, 0.53–0.77) or a mixed RCC histology (n = 85; median PFS, 5.7 vs 2.9 months; HR, 0.60; 95% CI, 0.33–0.85). Patients aged 65 years or older (n = 239; HR, 0.77; 95% CI, 0.58–1.03) and those younger than 65 years (n = 410; HR, 0.54; 95% CI, 0.43–0.68) had significant improvement in PFS, indicating that age did not affect the response to PFS. In addition, PFS did not seem to be affected by reduced kidney function, as assessed through creatinine clearance (CCr) or VEGF levels. Patients with both high/normal CCr (n = 131) or low CCr (n = 191) benefited from bevacizumab plus interferon (HR, 0.60; 95% CI, 0.46–0.79 and HR, 0.65; 95% CI, 0.51–0.82, respectively). Baseline VEGF levels were established based on recruitment, and improvements in PFS were observed in patients with VEGF levels below the median baseline level (HR, 0.44; 95% CI, 0.32–0.64) and above the median level (HR, 0.66; 95% CI, 0.49–0.93).
The tolerability profile for bevacizumab plus interferon in the AVOREN trial was consistent with the side effects previously reported for both agents. The dose intensity (percentage of planned total dose) of bevacizumab/placebo and interferon was similar in the two arms (92% bevacizumab plus interferon vs 96% interferon plus placebo for the bevacizumab/placebo arms, and 91% bevacizumab plus interferon vs 96% interferon plus placebo for the interferon arms). The incidence of grade 3/4 events associated with bevacizumab therapy included hypertension (7%), proteinuria (4%), bleeding (3%), arterial and venous thromboembolic events (3%), gastrointestinal perforation (1%), and wound-healing complications (<1%). In the final report, overall survival was not significantly improved (23.3 vs 21.3 months; unstratified HR, 0.91; 95% CI, 0.76–1.10; P = .3360; stratified HR, 0.86; 95% CI, 0.72–1.04; P = .1291). However, a trend favoring the combined treatment was reported.
Cancer and Leukemia Group B 90206 Trial
The Cancer and Leukemia Group B (CALGB) 90206 trial was the second major randomized open-label phase III trial to compare the efficacy and safety of bevacizumab plus interferon against interferon alone in patients with mRCC (n = 732). This study differed from the AVOREN study in that it was not placebo-controlled or blinded. The results from this trial confirmed the PFS data observed in the AVOREN trial, whereby the addition of bevacizumab to interferon improves PFS (median PFS for bevacizumab plus interferon was 8.5 months compared with 5.2 months for interferon alone; HR, 0.71; 95% CI, 0.61– 0.83). The PFS data for subgroups, including those based on MSKCC risk, were also confirmed. The phase III CALGB 90206 trial showed no new safety signals with the bevacizumab plus interferon regimen ( Table 1 ). Similar to the AVOREN trial, no differences in overall survival were seen between the combined arm and the interferon-alone arm.
| Treatment | Line | No. of Patients | Response Rate | PFS (mo) | OS (mo) |
|---|---|---|---|---|---|
| Anti-VEGF | |||||
| AVOREN Bevacizumab + interferon versus interferon + placebo | First | 649 | PR: 31% versus 13% CR: 1% | 10.2 versus 5.4 HR: 0.63 | 23.3 |
| CALGB 90206 Bevacizumab + interferon versus interferon | First | 732 | PR: 25.5% versus 13% CR: <1% | 8.5 versus 5.2 HR: 0.71 | 18.3 |
Bevacizumab combined with interferon received an approval as a first-line treatment for patients with advanced or metastatic RCC by the European Medicines Agency in December 2007 and the U.S. Food and Drug Administration (FDA) in July 2009.
Bevacizumab-based combination regimen
Bevacizumab, combined with a second targeted agent, has been evaluated with either tyrosine kinase inhibitors or mammalian target of rapamycin (mTOR) inhibitors ( Table 2 ).
| Second Agent | Reference, Phase I Trial | Ongoing Phase II–III Combination Trials | |
|---|---|---|---|
| Bevacizumab + tyrosine kinase inhibitor | |||
| Bevacizumab | Sunitinib | Feldman et al, 2009 | – |
| Bevacizumab 5 mg | Sorafenib (200 mg) | Sosman et al, 2008 | BeST (NCT00378703) |
| Bevacizumab + mTOR inhibitor | |||
| Bevacizumab, 10 mg | Temsirolimus, 25 mg | Merchan et al, 2009 | TORAVA (NCT00619268) INTORACT (NCT00631371) |
| Bevacizumab | Everolimus | Hainsworth et al, 2010 | RECORD-2 (NCT00719264) |
From the rationale of dual inhibition of the VEGF and mTOR pathways in RCC, phase I studies have been performed that have shown the feasibility of combining bevacizumab with one of the mTOR inhibitors, temsirolimus or everolimus. Based on preliminary encouraging data, several randomized trials have been designed.
The TORAVA phase II trial was a three-armed combination trial of bevacizumab plus interferon (n = 40) versus bevacizumab plus temsirolimus (n = 80) versus sunitinib (n = 40). The primary objective was nonprogression of RCC at 48 weeks. However, this trial reported a high frequency of grade 1 to 3 adverse events, especially anal fistulization. These combinations were discontinued in 41% of patients in the investigational arm because of toxicity. In terms of efficacy, the results were negative, with a median PFS of 8.2 months in the experimental arm compared with 16.8 and 8.2 months in the bevacizumab plus interferon and sunitinib arms, respectively. A phase III trial (INTORACT) comparing this combination with a combination of bevacizumab plus interferon was completed and should be reported shortly.
The combination of bevacizumab plus everolimus either as first-line treatment or after treatment with sunitinib or sorafenib in patients with advanced clear cell RCC was evaluated in a phase II trial. A total of 80 patients were enrolled in the trial. All patients received bevacizumab, 10 mg/kg intravenously every 2 weeks, and everolimus 10 mg, orally daily; patients with an objective response or stable disease continued treatment until disease progression or unacceptable toxicity occurred. Median PFS in patients who were treatment-naïve (n = 50) was 9.1 months versus 7.1 months in those previously treated (n = 30). Overall response rates were similar in both groups. Generally the combination regimen was well tolerated and, except for grade 3 to 4 proteinuria (25%), which led to treatment discontinuation in six patients, the toxicity profile was as expected. Despite the promising antitumor activity and good safety profile of this combination regimen, further studies are needed to compare it with sequential use of these two agents. A phase III CALGB study is investigating this combination versus everolimus alone in patients for whom prior VEGF-targeted therapy failed. The phase II trial (RECORD 2) investigating the combination of bevacizumab plus everolimus versus bevacizumab plus interferon (N = 360) has completed accrual and results are expected in 2012.
Bevacizumab in a neoadjuvant setting
Little is known about the use of bevacizumab in the neoadjuvant setting for RCC. Several retrospective analyses of perioperative complications in patients with mRCC, who had undergone cytoreductive nephrectomy after receiving various antiangiogenic agents, did not report excessive morbidity. One phase II trial assessed the feasibility of bevacizumab after four cycles as a neoadjuvant in 50 patients, but wound dehiscence resulted in treatment discontinuation for three patients and treatment delay for two others. Primary tumor regression of greater than 10% was observed in 10 of the 45 evaluable patients.
VEGF Trap
VEGF Trap (Regeneron Pharmaceuticals, Tarrytown, New York and sanofi-aventis, Bridgewater, New Jersey) is a fusion compound composed of the human VEGFR-1 (Flt-1) extracellular immunoglobulin domain number two and the VEGFR-2 (KDR) extracellular immunoglobulin domain number three, fused to the human IgGg1 Fc molecule. Therefore, this fusion protein acts as a soluble decoy receptor to bind VEGF and prevent subsequent VEGF binding and signaling. VEGF Trap binds to VEGF with a great affinity (Kd = 1 pmol/L) and also binds the placental growth factor, another angiogenic protein. In cultured endothelial cell assays, VEGF Trap showed inhibition of VEGF-induced VEGFR-2 phosphorylation and endothelial cell proliferation. In xenograft models, mice treated with VEGF Trap exhibited significant growth inhibition of different tumor subtypes. VEGF Trap activity has been assessed in phase I trials. In two trials, patients presented with refractory solid tumors. In the first report, 38 patients, including 9 with mRCC, received one or two subcutaneous doses of VEGF Trap, followed 4 weeks later with 6 weekly injections (escalating dose levels of 0.025, 0.05, 0.1, 0.2, 0.4, and 0.8 mg/kg) or six twice-weekly (0.8 mg/kg) injections. Drug-related grade 3 adverse events included hypertension and proteinuria, although a maximum tolerated dose was not determined. No anti–VEGF Trap antibodies were detected. No objective responses were observed in this trial. Of the 24 assessable patients, 14, including 5 of 6 at the highest dose level, maintained stable disease for 10 weeks. In the second trial, 30 patients were treated with intravenous VEGF Trap every 2 weeks at one of five different dose levels (0.3, 1.0, 2.0, 3.0, and 4.0 mg/kg). Drug-related grade 3 adverse events included arthralgia and fatigue. One patient with mRCC maintained a stable disease for more than 11 months (at the 1.0 mg/kg dose level). Dynamic contrast-enhanced magnetic resonance vascular imaging performed at baseline and after 24 hours indicated effective inhibition of tumor perfusion at the higher dose levels (2.0 mg/kg). Complete binding of circulating VEGF was documented at higher dose levels (2.0 mg/kg), with more free than bound VEGF Trap observed in the plasma. Further investigation is ongoing through an Eastern Cooperative Oncology Group (ECOG) phase II trial (ECOG-E4805, NCT00357760) that randomized 120 patients with mRCC to two different doses of VEGF Trap, with a primary end point of PFS at 8 weeks.
Ramucirumab (IMC-1121B; ImClone Systems Corporation, Branchburg, New Jersey) is a human mAb that specifically inhibits VEGFR-2; which is a critical receptor involved in malignant angiogenesis. Multiple clinical trials have been conducted to investigate the antitumor activity of ramucirumab in a variety of tumor types, such as RCC, colon cancer, non–small cell lung cancer, and hepatocellular carcinoma. A phase II study was recently presented of treatment with ramucirumab after tyrosine kinase inhibitors failed in patients with mRCC. Among 40 patients enrolled in the trial, 54% had received prior sunitinib, 10% received prior sorafenib, and 36% received both sunitinib and sorafenib. Patients received ramucirumab, 8 mg/kg, intravenously every 2 weeks. Overall response rate was 5%, and 38% of patients had stable disease. The preliminary median was 8.3 months, with a median follow-up of more than 1 year.
Common toxicities were headache, fatigue, epistaxis, peripheral edema, nausea, and dyspnea. Serious adverse events included grade 2 proteinuria and grade 2 hemoptysis in a patient with endobronchial metastases. Grade 3 or 4 adverse events occurred in 23% of patients and included grade 4 myocardial infarction and grade 3 syncope, hypertension, fatigue, dyspnea, sensory neuropathy, headache, back pain, polyneuropathy, decreased hemoglobin, and anorexia. Grade 4 cardiopulmonary arrest followed by death 13 months after the initiation of study therapy was reported in two patients with underlying cardiovascular disease. These results suggested that ramucirumab could have clinical activity as second- or third-line treatment in patients with mRCC refractory to tyrosine kinase inhibitors.
VEGF binding agents
Bevacizumab
Bevacizumab (Avastin, Genentech, South San Francisco, CA, USA) is a recombinant monoclonal antibody (mAb) IgG1 antibody that has been developed for humans from murine anti-VEGF mAb A4.6.1. The murine mAb A4.6.1 is specific for human VEGF, binding to all of the known isoforms of the ligand (eg, VEGF121, VEGF165, VEGF181, VEGF206). It is formed through alternative gene splicing, preventing it from binding to VEGFRs on vascular endothelial cells. In 1997, murine anti-VEGF mAb A4.6.1 was adapted for human use by site-directed mutagenesis, resulting in the production of bevacizumab. Bevacizumab is 93% human and 7% murine, and recognizes all of the major isoforms of human VEGF with a binding affinity of Kd = 8 × 10 −10 M (a similar affinity to the murine antibody). The binding ability of bevacizumab for VEGF is restricted to human, nonhuman primate, and rabbit VEGF. Sustained inhibition of VEGF with bevacizumab results in the regression of existing tumor microvasculature and normalization of surviving tumor vasculature, and inhibits the formation of new vasculature. It may also revert tumor-associated immune suppression and improve concomitant drug delivery into the tumor.
Bevacizumab has a terminal half-life of 17 to 21 days, with no dose-limiting toxicity when used as a single agent. The low interpatient variability and the modest effects of covariates on the clearance and volume distribution of bevacizumab support the current strategy of dosing bevacizumab based on body weight (mg/kg).
Phase II trials
Two key phase II trials have been conducted on bevacizumab use in treating RCC: AVF0890s and the RACE trial. The first trial, AVF0890s, was a randomized, placebo-controlled, double-blind trial of bevacizumab monotherapy conducted in patients with metastatic, predominantly clear cell RCC who were not optimal candidates for an interleukin (IL)-2 therapy or had not previously experienced response to this therapy. Between October 1998 and September 2001, 116 patients were randomized to one of three treatment arms: placebo (n = 40) or bevacizumab at either 3 mg/kg (n = 37) or 10 mg/kg (n = 39). This trial showed that the median time to progression was significantly longer for the 10-mg/kg bevacizumab arm than for the placebo arm (4.8 vs 2.5 months; hazard ratio [HR], 2.55; P <.001). The median time to progression for the 3-mg/kg bevacizumab arm was 3 months, and was not significantly greater than the placebo arm (HR, 1.26; P = .053). Four (10%) patients in the 10-mg/kg bevacizumab arm experienced partial responses of variable duration (6, 9, 15, and >39 months, respectively). This trial provided the rationale for the using 10 mg/kg of bevacizumab in treating RCC.
The phase II RACE trial evaluated bevacizumab in combination with erlotinib in patients with mRCC based on the rationale that VEGF has been implicated to have anti-epidermal growth factor receptor resistance. A randomized, double-blind, placebo-controlled trial was conducted at 21 sites in the United States. Eligible patients were enrolled from March 2004 through October 2004 to receive bevacizumab, 10 mg/kg, every 2 weeks, plus either erlotinib, 150 mg orally daily, or a placebo daily. The median progression-free survival (PFS) was not significantly improved by the addition of erlotinib to bevacizumab (8.5 months with bevacizumab plus placebo vs 9.9 months with bevacizumab plus erlotinib; HR, 0.86; 95% CI, 0.50–1.49). Furthermore, the addition of erlotinib to bevacizumab resulted in a similar overall response rate, which was 13% with bevacizumab plus placebo versus 14% with bevacizumab plus erlotinib. The addition of erlotinib to bevacizumab did not result in an improved duration of objective response (6.7 vs 9.1 months) or time to symptom progression (HR, 1.172; P = .5076). This efficacy has been the basis for testing bevacizumab in phase III trials.
Phase III trials
AVOREN trial
AVOREN was the pivotal phase III trial to evaluate the efficacy and safety of adding bevacizumab to interferon for treating mRCC. Bevacizumab was tested in combination with interferon to determine whether it would add efficacy to one of the standard treatments at the time of the trial’s design. Between June 2004 and October 2005, the trial enrolled 649 patients from 18 countries. Eligible patients had mRCC with a predominantly clear cell histology (>50% clear cells if mixed), underwent prior nephrectomy for primary RCC, a measurable or nonmeasurable disease according to Response Evaluation Criteria in Solid Tumors (RECIST), a Karnofsky performance status of at least 70%, and no proteinuria at baseline (<0.5 g of protein in a 24-hour urine collection). Patients were randomized on a 1:1 basis to receive interferon (9 million international units) three times weekly plus a placebo, or bevacizumab at 10 mg/kg every 2 weeks plus interferon. The final analysis of PFS, which was performed at the scheduled time point for overall survival, showed that it was significantly improved by the addition of bevacizumab to interferon, for 5.4 to 10.2 months (HR, 0.63; P = .0001). This finding represents an 89% improvement in median PFS with bevacizumab plus interferon. The addition of bevacizumab to interferon also improves the overall response rate compared with interferon plus a placebo (31% vs 13%). Analyses of patient subgroups suggested that the addition of bevacizumab to interferon improves PFS in all subgroups analyzed. Improvements in PFS were observed in both favorable (n = 180) and intermediate (n = 363) Memorial Sloan-Kettering Cancer Center (MSKCC) risk categories (median PFS, 12.9 vs 7.6 months; HR, 0.60; P = .0039, and median PFS, 10.2 vs 4.5 months; HR, 0.55; P <.0001, respectively). A significant improvement was not seen in patients in the poor MSKCC risk category (n = 54; median PFS, 2.2 months with bevacizumab plus interferon vs 2.1 months with interferon plus placebo; HR, 0.81; P = .457). An improvement in PFS was observed in patients receiving bevacizumab plus interferon who either had a clear cell RCC histology (n = 564; median PFS, 10.2 vs 5.5 months; HR, 0.64; 95% CI, 0.53–0.77) or a mixed RCC histology (n = 85; median PFS, 5.7 vs 2.9 months; HR, 0.60; 95% CI, 0.33–0.85). Patients aged 65 years or older (n = 239; HR, 0.77; 95% CI, 0.58–1.03) and those younger than 65 years (n = 410; HR, 0.54; 95% CI, 0.43–0.68) had significant improvement in PFS, indicating that age did not affect the response to PFS. In addition, PFS did not seem to be affected by reduced kidney function, as assessed through creatinine clearance (CCr) or VEGF levels. Patients with both high/normal CCr (n = 131) or low CCr (n = 191) benefited from bevacizumab plus interferon (HR, 0.60; 95% CI, 0.46–0.79 and HR, 0.65; 95% CI, 0.51–0.82, respectively). Baseline VEGF levels were established based on recruitment, and improvements in PFS were observed in patients with VEGF levels below the median baseline level (HR, 0.44; 95% CI, 0.32–0.64) and above the median level (HR, 0.66; 95% CI, 0.49–0.93).
The tolerability profile for bevacizumab plus interferon in the AVOREN trial was consistent with the side effects previously reported for both agents. The dose intensity (percentage of planned total dose) of bevacizumab/placebo and interferon was similar in the two arms (92% bevacizumab plus interferon vs 96% interferon plus placebo for the bevacizumab/placebo arms, and 91% bevacizumab plus interferon vs 96% interferon plus placebo for the interferon arms). The incidence of grade 3/4 events associated with bevacizumab therapy included hypertension (7%), proteinuria (4%), bleeding (3%), arterial and venous thromboembolic events (3%), gastrointestinal perforation (1%), and wound-healing complications (<1%). In the final report, overall survival was not significantly improved (23.3 vs 21.3 months; unstratified HR, 0.91; 95% CI, 0.76–1.10; P = .3360; stratified HR, 0.86; 95% CI, 0.72–1.04; P = .1291). However, a trend favoring the combined treatment was reported.
Cancer and Leukemia Group B 90206 Trial
The Cancer and Leukemia Group B (CALGB) 90206 trial was the second major randomized open-label phase III trial to compare the efficacy and safety of bevacizumab plus interferon against interferon alone in patients with mRCC (n = 732). This study differed from the AVOREN study in that it was not placebo-controlled or blinded. The results from this trial confirmed the PFS data observed in the AVOREN trial, whereby the addition of bevacizumab to interferon improves PFS (median PFS for bevacizumab plus interferon was 8.5 months compared with 5.2 months for interferon alone; HR, 0.71; 95% CI, 0.61– 0.83). The PFS data for subgroups, including those based on MSKCC risk, were also confirmed. The phase III CALGB 90206 trial showed no new safety signals with the bevacizumab plus interferon regimen ( Table 1 ). Similar to the AVOREN trial, no differences in overall survival were seen between the combined arm and the interferon-alone arm.
Related posts:
 Renal Cell Carcinoma
Renal Cell Carcinoma
 mTOR Inhibitors in Advanced Renal Cell Carcinoma
mTOR Inhibitors in Advanced Renal Cell Carcinoma
 Management of Treatment-Related Toxicity with Targeted Therapies for Renal Cell Carcinoma: Evidence-Based Practice and Best Pr actices
Management of Treatment-Related Toxicity with Targeted Therapies for Renal Cell Carcinoma: Evidence-Based Practice and Best Pr actices
 Clinical and Molecular Prognostic Factors in Renal Cell Carcinoma: What We Know So Far
The Role of Surgery in the Management of Early-Stage Renal Cancer
mTOR Inhibitors in Advanced Renal Cell Carcinoma
Clinical and Molecular Prognostic Factors in Renal Cell Carcinoma: What We Know So Far
The Role of Surgery in the Management of Early-Stage Renal Cancer
mTOR Inhibitors in Advanced Renal Cell Carcinoma
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


