Osteosarcoma and chondrosarcoma are the 2 most common malignant bone tumors. This review discusses the clinicopathologic features, recent preclinical developments, and targets currently being or to be validated in the clinic.
Key points
- •
Since the introduction of conventional adjuvant chemotherapy 3 decades ago, survival of patients with osteosarcoma has reached a plateau of efficacy at 60% to 65%; patients with metastases at diagnosis have a poor fate.
- •
Immune stimulation is one of the few treatment options that may improve survival of patients with osteosarcoma.
- •
Chondrosarcoma is resistant to conventional chemotherapy and radiotherapy, and currently there are no curative options for patients with inoperable or metastatic disease.
- •
Targeting of apoptosis and survival pathways are promising approaches that may improve survival of patients with chondrosarcoma.
Introduction
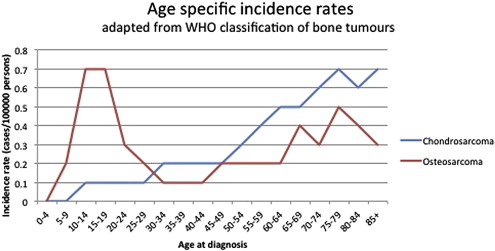
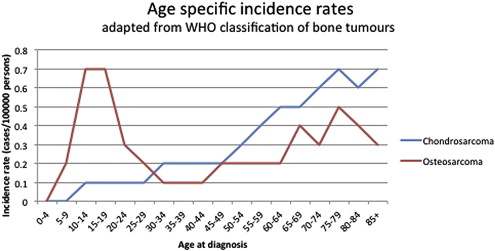
Primary bone tumors are rare and have a specific age distribution ( Fig. 1 ). Conventional osteosarcoma is the most frequent primary high-grade bone tumor in humans; there are 4 new cases per 10 6 population per year with the highest incidence in adolescence. The second most frequent primary bone malignancy, chondrosarcoma, accounts for approximately 3 new cases per 10 6 population per year predominantly affecting adults. The clinical management of unresectable and metastatic disease as well as therapy resistance remain a clinical challenge. This review discusses the molecular pathways that have been identified as a result of intensive genome-wide and basic biology analysis and the rationale for current clinical and preclinical targets for therapy for these 2 most frequent bone sarcomas.
Introduction
Primary bone tumors are rare and have a specific age distribution ( Fig. 1 ). Conventional osteosarcoma is the most frequent primary high-grade bone tumor in humans; there are 4 new cases per 10 6 population per year with the highest incidence in adolescence. The second most frequent primary bone malignancy, chondrosarcoma, accounts for approximately 3 new cases per 10 6 population per year predominantly affecting adults. The clinical management of unresectable and metastatic disease as well as therapy resistance remain a clinical challenge. This review discusses the molecular pathways that have been identified as a result of intensive genome-wide and basic biology analysis and the rationale for current clinical and preclinical targets for therapy for these 2 most frequent bone sarcomas.
Osteosarcoma
Clinicopathologic Features
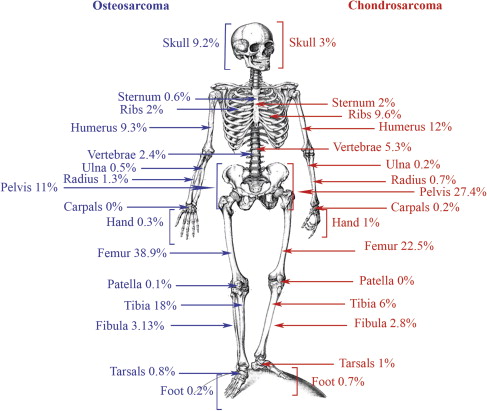
Conventional osteosarcoma is defined as a high-grade intraosseous malignant neoplasm in which the neoplastic cells produce bone. Osteosarcoma represents about 55% of all malignant bone tumors and occurs predominantly in children and adolescents (see Fig. 1 ). Most osteosarcomas are located around the knee (60%, Fig. 2 ) and other long tubular bones of the skeleton. Histologically, a broad spectrum of morphology can be seen, including varying amounts of osteoid, cartilage, and/or fibrous material. In addition to conventional osteosarcoma, the World Health Organization 2013 classification recognizes low-grade central, parosteal, periosteal, and high-grade surface osteosarcoma as well as telangiectatic and small cell osteosarcoma as distinct, less frequent subtypes ; the latter 2 are also of high histologic grade. All high-grade osteosarcomas have a similar clinical behavior with a high risk of metastasis.
Osteosarcoma is primary when the underlying bone is normal and secondary when the bone is altered by conditions such as previous radiation or Paget disease. There is an increased incidence of primary osteosarcoma associated with several genetic syndromes, eg, Li-Fraumeni syndrome (TP53), hereditary retinoblastoma (Rb), or Rothmund-Thomson (RECQ-helicase); in older adults it is often secondary. Although rare, the incidence of osteosarcoma in these syndromes is manifold higher than in the general population, suggesting that the genes involved in these syndromes play an important role in the biology of osteosarcomas. Dismal prognostic factors for osteosarcoma are metastatic disease at diagnosis, proximal or axial tumor site, large size, and poor histologic response to preoperative chemotherapy.
Current Management of Osteosarcoma and Resistance to Therapy
Osteosarcoma should be treated by an experienced multidisciplinary team and treatment consists of preoperative and postoperative chemotherapy and (complete) surgical resection of the tumor and, if present, metastatic tumor deposits. Despite intensive treatment strategies, the chance of cure in patients with resectable osteosarcoma has remained around 60% to 65% in the past 3 decades, indicating that a plateau of efficacy regarding conventional chemotherapy has been reached. In nearly all failures of treatment, the patients die from systemic spread of the disease, mainly to the lungs and bones. Resistance to cytotoxic drugs has been recognized in osteosarcoma, and is a marker for poor prognosis. However, patients with a good histologic response to chemotherapy may also develop metastases or recurrent disease, suggesting the presence of heterogeneous tumor cell populations. There is cumulating evidence that osteosarcomas contain cells with a tumor-initiating or so-called stem cell phenotype displaying a high tumorigenic capacity, and harboring multidrug resistance proteins contributing to resistance and recurrence of disease.
Targets and Novel Treatment Options in Osteosarcoma
Osteosarcoma is characterized by a highly instable and complex karyotype, suggesting that genetic instability is the initial cause of osteosarcoma genesis. The recently described phenomenon chromothripsis, indicating scattering of chromosomes, is typical in osteosarcoma. However, repair of genetic instability does not restore the damaged done. Several molecular pathways involved in osteosarcoma genesis are discussed as far as these are relevant and druggable ( Table 1 ).
| Target | Drug | Mechanism | Type of Trial and Clinical Results (n = Number of OS Cases) | Reference or Clinical Trial Identifier |
|---|---|---|---|---|
| IGF-1R | SCH717454/RG1507 | MoAb anti-IGF-1R | Phase 1 (n = 3), NOR | |
| EGF/EGF-R | Gefitinib | Small anti-EGF-R TK | Phase 1 (n = 9), NOR | |
| PDGF/PDGF-R; KIT | Erlotinib | Small anti-EGF-R TK | Phase 1 (n = 6), NOR | |
| Imatinib/STI571 | Small anti-PDGF-R TK | Phase 2 (n = 38), NOR | ||
| Sorafenib | Small anti-VEGFR, anti-PDGFR and anti-KIT TK | Phase 2 (n = 35), 3 PR | ||
| Pazopanib | Small anti-VEGFR1, -2, -3 PDGFRα/β and anti-Kit TK | NCT01759303 | ||
| HER2 | Trastuzumab | MoAb anti-HER2 | Phase 2 (n = 41 HER2+), no difference in OAS/EFS between HER2+ and − | |
| VEGF/VEGFR | Bevacizumab (+sorafenib/cyclo) | MoAb against 4 VEGF isoforms | Phase 1 (n = 5), NOR | |
| Endostatin | MoAb against VEGFR2 | NCT01002092 | ||
| Cediranib/AZD2171 | Small anti-VEGFR1, -2, -3 and anti-Kit TK | Phase 1 (n = 2), 1 PR | ||
| DNA synthesis | Gemcitabine | Nucleoside analogue | Phase 2 (n = 10), NOR | |
| Gemcitabine + docetaxel | Nucleoside analogue | Phase 2 (n = 30), 6 PR | ||
| Pemetrexed (PMX) | Folate antagonist | Phase 2 (n = 32), 1 PR; phase 2 (n = 10), NOR | ||
| DNA minor groove | Ecteinascidin-743 | Alkylating agent, inhibits cell cycle progression, enhances apoptosis | Phase 1 (n = 7), 2 PR; phase 2 (n = 23) NOR | |
| Microtubules | Paclitaxel | Enhances microtubule polymerization, disrupts microtubule network, induces apoptosis | Phase 2 (n = 11), NOR | |
| Docetaxel | Phase 2 (n = 34), 1 CR, 2 PR | |||
| Ixabepilone | Phase 2 (n = 11), NOR | |||
| RANK/RANKL | Zoledronic acid | Blocks mevalonate pathway, preventing isoprenylation of GTP-binding proteins, small, membrane-bound G-proteins cannot function | NTC00691236 | |
| Pamidronate | See zoledronic acid | Phase 2 (n = 29), no efficacy reported, no serious side effects | ||
| SRC | Saracatinib | Inhibits tumor-induced SRC overexpression osteoclasts | NTC00752206 | |
| AKT/PI3K/mTOR | Deforolimus | mTOR inhibitor | Phase 1 (n = 1), NOR | |
| Ridaforolimus | mTOR inhibitor | Phase 2 (n = 212) 2 OS patients showed PR | ||
| Sirolimus + cyclophosphamide | Antiproliferative, cell cycle arrest, antiapoptotic | Phase 2 (n = 6), NOR | ||
| Everolimus | Binds to mTORC1 complex, inhibits downstream signaling; inhibits VEGF and HIF | NCT01216826 | ||
| Multiple | Curcumin | Cell cycle arrest in G2/M, increased apoptosis (decrease Bcl-2, increase Bax IC), inhibition migration/adhesion. Antagonizing growth factors, protein kinases, cytokine signaling, cyclin D, and upregulates p53 | NCT00689195 | |
| INF-α/β | INF-α/β | Activation of Ifn type 1–specific response elements via the TK/JAK-STAT1/2 pathway, activating T cells, macrophages, and NK cells | Phase 2 (n = 20), 4 PR; phase 3 (n = 89), 10y-EFS 39% | |
| Immune-competent cells | MTP-PE | MTP binds NOD2 receptor in monocytes, dendritic cells, and macrophages leading to NF-κB activation | Phase 2 (n = 28), TTR 9.0 vs 4.5 mo ( P <.03); phase 3 (OS IIb, n = 338), OAS 78% vs 70% ( P <.03); phase 3 (OSIII, n = 46), OAS 53% vs 40% ( P = .27) | |
| GM-CSF | Sargramostim | Increase number/cytotoxicity of macrophages, T cells, NK cells, and dendritic cells | Phase 2 (n = 43); NOR | |
| GM-CSF + gemcitabine | Increase immunologic response and FAS expression | |||
| T cell | 105AD7 vaccine | Immune response against CD55 | Phase 2 (n = 14), n = 5 have response | |
| Anti-GD2 | Immune response against disialoganglioside (GD2) | Phase 1 (n = 1), NOR | NCT00743496 |
P53-related signaling
Mutational inactivation of the tumor suppressor gene TP53 disrupts checkpoint responses to DNA damage, resulting in the potential for destabilization of the genome. TP53 mutations cause Li-Fraumeni syndrome; somatic TP53 mutations also occur in 15% to 30% of osteosarcomas. Presumed associations with prognosis are not reproducible. MDM-family proteins that are involved in p53 protein degradation have been shown to be upregulated in many tumor types, although amplification of MDM2 in osteosarcoma was especially found in the parosteal subtype. Recently, an association with good prognosis and high expression of an MDM4 splice variation has been shown in osteosarcoma. Both MDM2 and MDM4 are druggable targets through Nutlin-3 ; RO5045337, and other drugs are in development ( NCT01164033 ).
Retinoblastoma signaling
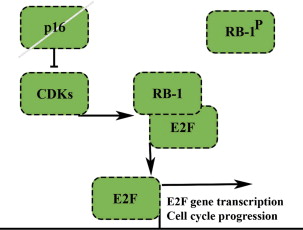
The tumor suppressor retinoblastoma gene RB1 has a central role as a checkpoint in the cell cycle ( Fig. 4 ). Loss of function as occurring in patients with an RB1 germline mutation increases the risk of osteosarcoma up to 500-fold. In osteosarcomas, RB1 gene abnormalities have been found in 26% to 53% of patients. Inactivation of the RB1 gene also contributes to genomic instability in osteosarcomas.
The product of the CDKN2A/p16 gene is part of the Rb pathway inhibiting cell cycle progression by preventing the formation of the cyclin D-Cdk4 complex. Genomic loss, rather than inactivation by hypermethylation of CDKN2A/p16 was strongly associated with a poor prognosis in osteosarcoma. Homozygous deletion of the entire CDKN2 locus, including p16 , p19 , and p15 is always a recurrent event in oncogenic transformation of mouse mesenchymal stem cells, which form osteosarcomalike lesions on transplantation into mice. However, only 10% of human osteosarcomas harbor a CDKN2A homozygous deletion.
Pharmacologic intervention to restore the defective RB signaling does not seem feasible. Identification of homozygous deletion of CDKN2A may serve as a useful prognostic marker to identify tumors with a very poor prognosis.
Developmental pathways: Wnt, TGF, bone morphogenetic protein, and hedgehog
Canonical Wnt signaling, through nuclear β-catenin, is important in many developmental processes, including osteogenic differentiation and many different cancer types. Both upregulation and downregulation of the Wnt/β-catenin pathway has been reported in osteosarcoma. Restoration of Wnt/β-catenin activation resulted in decreased proliferation and differentiation of the tumor cells, thereby suggesting a tumor suppressive role for this pathway in osteosarcoma.
The noncanonical or β-catenin independent Wnt pathway has also been implicated in osteosarcoma, predominantly via the receptor tyrosine kinase ROR2. The Wnt5a ligand and ROR-2 receptor contribute to invasiveness of osteosarcoma cells, mediated via the matrix-metalloproteinase, MMP13. Because the receptor tyrosine kinase ROR2 was found to be overexpressed in osteosarcomas, inhibition of this receptor could be a potential target. Clinical phase 1 studies targeting the Wnt pathway are under way, such as the Wnt-pathway antagonist OMP-54F28 ( NCT01608867 ).
Other developmental pathways in osteogenesis include the SMAD-mediated TGFβ and bone morphogenetic protein pathways and the hedgehog pathways. Decreased expression of phosphorylated SMAD2, indicating decreased TGFβ signaling, was shown to be associated with poor survival in osteosarcoma. Activity of the hedgehog pathway is variable in osteosarcoma and cyclopamine, an inhibitor of hedgehog signaling was shown to inhibit osteosarcoma cell line proliferation, but this was independent of hedgehog signal transduction and could most likely be attributed to a specific toxicity of this compound. Other drugs targeting the hedgehog pathway are currently under development in (osteo)sarcoma; eg, IPI-906, LDE225, and GDC-0449.
Proliferative pathways: insulin growth factor
In vertebrates, insulinlike growth factor 1 (IGF-1) is a major growth-promoting signal for skeletal development. Aberrant signaling of the IGF-1 pathway through its receptors IGF-1R and IR (insulin receptor) has been implicated in various cancer types, including sarcomas. Treatment with different monoclonal antibodies against IGF-1R has been performed in xenograft models of osteosarcoma, in which a response was detected in at least 60% of all cases studied.
Proliferative pathways: platelet-derived growth factor
Platelet-derived growth factor-α (PDGF-AA) and its receptor (PDGFRA) were found to be expressed on osteosarcoma cells, suggesting an autocrine/paracrine loop of growth stimulation, although mutations were absent. Inhibition of PDGFRA using imatinib in osteosarcoma cell lines blocked intracellular PDGFRA-induced cell signaling, but the downstream target, mitogen-activated protein kinase, was constitutively activated, indicating an escape route of induction. Expression of KIT, another tyrosine kinase receptor blocked by imatinib, was found in 20% of osteosarcomas, although KIT mutations were absent. The multi (including PDGFRA) tyrosine kinase inhibitor sorafenib was found to be active in preclinical osteosarcoma models and in a phase 2 study. These results warrant further studies of this (class of) drugs in osteosarcoma.
Proliferative pathways: human epidermal growth factor
The human epidermal growth factor receptor 2 (HER2) is a 185-kDa membrane-bound glycoprotein, encoded by the ErbB2 protooncogene. Overexpression of the HER2 gene by ErbB2 amplification results in oncogenic transformation of cells. In osteosarcomas, HER2 overexpression is associated with metastasis and with increased expression of P-glycoprotein. Targeted treatment with the monoclonal antibody trastuzumab is an effective treatment in HER2-overexpressing breast cancers, and is supposed to have a beneficial effect in osteosarcomas. Therefore, a phase 2 trial in patients with osteosarcomas has been launched. However, not all groups could confirm HER2 overexpression in osteosarcoma, and the theoretic backbone for the use of trastuzumab is debatable. The phase 2 trial did not show a difference in outcome among presumed HER2+ and HER2− patients, suggesting that HER2 should not be regarded as a target for treatment of osteosarcoma.
Immune modulation
The role of immunotherapy in osteosarcoma has not been elucidated yet, but has been regarded as a magic box since the use of Coley’s toxin, which produced a regression in osteosarcomas that were not amenable to surgery. The recent insights in the complex immunologic interplay of immune-competent cells in cancer, the plasticity of this immune response, and diverse effects of infiltrating macrophages in carcinomas versus sarcomas have increased the interest in immunomodulation of osteosarcomas. It has been shown that the presence of tumor-infiltrating macrophages in biopsy samples of osteosarcoma was associated with reduced metastatic propensity and better survival. This observation could be a rationale for adjuvant treatment with muramyl tripeptide phosphatidylethanolamine (MTP-PE), an immune stimulatory drug. However, the results of the landmark phase 3 study investigating the benefit of MTP-PE in frontline treatment of osteosarcoma has been a matter of debate, leaving the indication for this drug unsure or unregistered in many countries.
In addition, the cytotoxic potential of natural killer cells against osteosarcoma cells was demonstrated to be activated by IL15 or cetuximab. Other drugs that can activate macrophage activity in tumors that have not been investigated yet in osteosarcoma, such as paclitaxel, are reported to activate the macrophage response in vitro, through a mechanism similar to bacterial liopopolysaccharides.
Immune stimulation by interferon (Ifn) was supported by in vitro studies, demonstrating a more intensive effect of Ifn-α compared with the type-2 Ifn, Ifn-γ. Nonrandomized clinical experience with Ifn-α showed a long-term event-free survival of 40% in a patient group treated with Ifn-α as adjuvant to local treatment with surgery. Limited clinical experience had shown a transient effect of Ifn-α in patients with lung metastases and advanced disease. The recently closed EURAMOS-1 study will provide a more definitive answer about the contribution of immune modulation later in 2013, as this study included an arm with Ifn-α in addition to conventional chemotherapy in good responders after primary osteosarcoma treatment ( www.euramos.org ).
Other immunologic approaches, for example, inhalation of granulocyte-macrophage colony-stimulating factor (GM-CSF), did not seem effective in patients, and addition of interleukin-1α to etoposide has an unwarranted spectrum of side effects, too serious to use on a large scale in patients. Vaccination with the antiidiotype vaccine 105AD7 showed measurable T-cell responses in patients, but the advantage for patients with osteosarcoma in the clinic needs to be established.
Other therapies: bisphosphonates and tyrosine kinase inhibitors
Bisphosphonates such as zoledronic acid or pamidronate inhibit osteoclast activity and are found to be effective in avoiding complications caused by bone metastases. However, (pre)clinical experience with these compounds proved that bisphosphonates also exerted antineoplastic effects. In osteosarcoma cells, a caspase-independent apoptosis was reported after exposure to zoledronic acid. This P53-independent apoptosis occurred via mitochondrial pathways in these osteosarcoma cells. In another study, the presence of the drug-resistance protein P-glycoprotein did not impair the anticancer activity of the bisphosphonates. In addition, osteosarcoma cells could be inhibited in their migration capacity and lung metastases could be suppressed by bisphosphonates. These antitumor effects have led to the initiation of a phase 2 clinical trial with pamidronate in addition to the routine chemotherapy schedule for patients with localized or metastatic osteosarcoma. Event-free survival in this uncontrolled trial for patients with localized disease after 5 years was 72% and overall survival was 93%, whereas 45% of the patients with metastatic disease survived without recurrence. Currently, the French led OS2006 trial is investigating the addition of pamidronate to frontline chemotherapy in patients with resectable osteosarcoma in a randomized fashion.
Chondrosarcoma
Clinicopathologic Features
Chondrosarcomas are hyaline cartilaginous tumors most often arising in bones that develop during endochondral ossification. Incidence and location are shown in Figs. 1 and 2 . Conventional chondrosarcoma accounts for approximately 85% of all primary chondrosarcomas and prognosis is strongly correlated with histologic grading. Grade I chondrosarcoma, now reclassified as an atypical cartilaginous tumor, shows low cellularity and is locally aggressive, but typically does not metastasize. Grade II and grade III conventional chondrosarcomas show increased cellularity with mitoses and reduced cartilaginous matrix, and a corresponding increase in metastasizing capacity alongside poor patient survival. Amongst the rare chondrosarcoma subtypes, dedifferentiated chondrosarcoma accounts for up to 10% of all chondrosarcomas and has a dismal prognosis. Dedifferentiated chondrosarcoma is composed of 2 histologically distinct components: a high-grade dedifferentiated component and a seemingly low-grade cartilaginous component. Mesenchymal chondrosarcoma is considered high grade and accounts for approximately 3% of primary chondrosarcomas, histologically showing undifferentiated small round cells admixed with well-differentiated cartilage. Clear cell chondrosarcoma is considered low grade and accounts for 2% of all primary chondrosarcomas, demonstrating tumor cells with a clear empty cytoplasm. Extraskeletal myxoid chondrosarcoma is primarily located in soft tissue and is considered a misnomer because it lacks cartilaginous differentiation, and is therefore out of the scope of this review.
Current Management of Chondrosarcoma and Resistance to Therapy
The firstline treatment of chondrosarcoma is surgical resection with local adjuvant treatment such as phenol or cryosurgery, followed by filling the cavity with bone graft, showing long-term local control in atypical cartilaginous tumor/grade I chondrosarcoma. Because of the necessity for wide resection margins to prevent recurrence in grade II and III chondrosarcomas, the patient often needs to undergo mutilating surgery. In the event of tumor location at a nonresectable site, such as in the skull or pelvis, or metastatic disease, there is still no curative treatment. Chondrosarcoma is notorious for its resistance to conventional chemotherapy and radiotherapy. Recently, a phase 2 study including 25 patients with chondrosarcoma using the nucleoside analogue gemcitabine (657 mg/m 2 on day 1 and day 8) followed by the antimitotic docetaxel (75 mg/m 2 on day 8) over a course of 21 days was aborted because only 2 patients showed partial response. In a recent study including 9 patients with dedifferentiated chondrosarcoma treated with surgery and chemotherapy (adriamycin, ifosfamide, cisplatin, and methotrexate), all patients died of metastatic disease. These results illustrate the need for new targeted treatments in chondrosarcoma, because the conventional chemotherapeutics targeting the DNA machinery are not effective.
Primary chemoresistance of chondrosarcoma has long been ascribed to the phenotypic properties, such as hyaline cartilaginous matrix surrounding the cells prohibiting access to the cells, poor vascularization, and a slow division rate. As these properties are less prominent in high-grade chondrosarcomas, which typically show less matrix, increased vascularization, and increased mitotic rate, the resistance to therapy could also be due to activated antiapoptosis or prosurvival pathways. Moreover, nuclear accumulation of doxorubicin was shown despite the presence of matrix and multidrug resistance pump activity. In addition, inhibition of the antiapoptotic Bcl-2 family members was found to overcome resistance to doxorubicin and cisplatin in chondrosarcoma cell lines.
Targets and Novel Treatment Options in Chondrosarcoma
In recent years, advances have been made in identifying multiple active pathways in chondrosarcoma, and preclinical work has led to the identification of potential targets for clinical trials ( Table 2 ). Here, the recent identification of IDH mutations is discussed in relation to active survival pathways and HIF1α expression found in high-grade chondrosarcomas, as well as growth plate signaling pathways including antiapoptotic signaling, and retinoblastoma pathway alterations.
| Target | Drug | Mechanism | Clinical Results | Clinical Trial Identifier or Reference |
|---|---|---|---|---|
| DNA synthesis | Gemcitabine | Nucleoside analogue | Phase 2 (n = 53) combination with docetaxel. Terminated due to lack of evidence of efficacy | |
| Permetrexed | Prevents formation of DNA and RNA | Study completed, no results posted | NCT00107419 | |
| AKT/PI3K | Perifosine | Inhibits AKT membrane recruitment | Phase 1 (n = 10) combination with gemcitabine Patient with CS showed 17% decrease in tumor size after 2 cycles | NCT00401388 (Steinert CTOS 2006) |
| mTOR | Sirolimus | mTOR inhibitor | Combination with cyclophosphamide in 10 patients Disease control rate of 70% | |
| SRC | Dasatinib | Small molecule kinase inhibitor | Phase 2, ongoing, NOR in CS | NCT00464620 (Schuetze CTOS 2006) |
| PDGF | Sunitinib (SU11248) | Multitargeted receptor tyrosine kinase inhibitor | Phase 2, completed, no results posted Case study: Antitumor activity in 2 patients with extraskeletal myxoid CS Case study: Durable response after combination with proton beam radiation in 1 patient with metastatic clear cell CS | NCT00474994 , |
| Imatinib | Competitive tyrosine kinase inhibitor | Phase 2 (n = 26), NOR | ||
| Pazopanib | Blocks autophosphosphorylation of PDGF receptors, VEGF receptors, FGF receptors 1 and 3; inhibits Kit and Lck | Recruiting | NCT01330966 | |
| Hedgehog | Saridegib (IPI-926) | Smoothened inhibitor | Study terminated due to lack of evidence of efficacy | NCT01609179 |
| Ongoing | NCT01310816 | |||
| Vismodegib (GDC-0449) | Smoothened inhibitor | Ongoing | NCT01267955 | |
| Apoptosis | Dulanermin rhAPO2L/TRAIL (AMG 951) | Induces apoptosis through binding to DR4 and DR5 | Phase 1 study (n = 71) 2 patients with CS durable PR Case study: near CR over 78 mo in 1 patient with metastatic disease | |
| PRO95780 | Monoclonal IgG1 antiantibody that triggers extrinsic apoptotic pathway through DR5 | Phase 1 study (n = 50), terminated due to lack of evidence of efficacy Patient with CS 20% reduction in measurable disease | ||
| Rb pathway | Alvespimycin | HSP90 inhibitor | Phase 1 study (n = 25) CS patient CR with reduction in CDK4 levels |
Survival pathways: isocitrate dehydrogenase mutations
Mutations in the isocitrate dehydrogenases (IDH) are found in 87% of benign enchondromas, 38% to 70% of primary conventional central chondrosarcomas, and 54% of dedifferentiated chondrosarcomas, but not in clear cell or mesenchymal chondrosarcomas. IDH is involved in the tricarboxylic acid cycle (Krebs cycle) and mutations in IHD1/2 lead to a diminished capacity to convert isocitrate to α-ketoglutarate (α-KG) and an acquired ability to convert αKG to d -2-hydroxyglutarate (D2HG), which is considered an oncometabolite.
The exact mechanism by which D2HG causes tumor formation is unknown although increasing evidence points toward epigenetic mechanisms. D2HG impairs the function of the αKG-dependent dioxygenase TET2, leading to inhibition of DNA demethylation causing CpG island hypermethylation. Enchondromas carrying IDH mutations were hypermethylated. In addition, D2HG was shown to impair histone demethylation. Moreover, mutations in IDH are postulated to inhibit the prolyl/lysyl/hydroxylation of collagen proteins and thereby their maturation as an IDH1 R132H conditional knock-in mouse model showed a reduction in collagen IV maturation. D2HG was postulated to induce pseudohypoxia ( Fig. 3 ) by inhibition of the HIF proline hydroxylases although this is controversial.
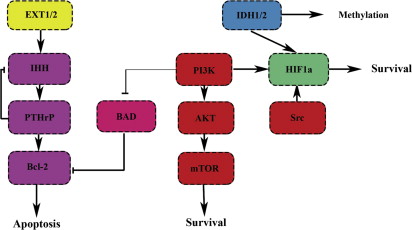
HIF-1α is upregulated by a multitude of malignancies to cope with reduced perfusion, and is associated with increased proliferation, vascular endothelial growth factor (VEGF) production, and resistance to chemotherapy and radiotherapy. High-grade conventional chondrosarcomas show activation of the hypoxia pathway through HIF1α. Most drugs targeting hypoxia, are designed either to target VEGF, the downstream target of HIF1α, or the phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT)/mammalian target of rapamycin (mTOR) pathway, which can induce HIF1α independent of oxygen conditions (see Fig. 3 ).
Survival pathways: PI3K, AKT, mTOR, VEGF
The PI3K/AKT pathway is often upregulated in cancer and can either inhibit apoptosis or promote cell proliferation (see Fig. 3 ). Active AKT signaling was shown in chondrosarcoma and the PI3K/AKT pathway has been shown to be involved in proliferation in mesenchymal chondroprogenitor cells. In chondrocytes, the PI3K/AKT can be activated by the chondrogenic transcription factor SOX9, which is also expressed in chondrosarcoma. SOX9 siRNA in a chondrosarcoma cell line (SW1353) induced apoptosis that could be rescued by phosphatase and tensin homolog (PTEN) expression. Mutations in the tumor suppressor PTEN are rare in chondrosarcoma. Perifosine, an AKT inhibitor inhibiting AKT membrane recruitment, showed a 17% decrease in tumor size in 1 patient with chondrosarcoma after 2 cycles (Steinert, CTOS 2006). A larger phase 2 study was conducted including patients with chemoinsensitive sarcomas but the results have not yet been posted ( NCT00401388 ).
mTOR is a point of convergence of many pathways involved in protein synthesis and cell proliferation, including the PI3K/AKT pathway (see Fig. 3 ). The first suggestion of activation of the mTOR pathway was in mesenchymal chondrosarcoma, showing strong cytoplasmic p-AKT, p-mTOR, and PDGFR-alpha staining. In an adjuvant rat orthotopic Swarm rat chondrosarcoma model, everolimus alone or in combination with doxorubicin after curettage showed inhibition of mTORC1 and decreased cell proliferation, however, the combination with doxorubicin showed an antagonistic effect with activation of the mTORC2 pathway. Allosteric inhibitors of the mTOR pathway, rapalogs (rapamycin [sirolimus], everolimus, and temsirolimus), have limited efficacy in the clinic, but show high synergy with dual PI3K/mTOR inhibitors such as BEZ235. A clinical trial with temsirolimus and liposomal doxorubicin included patients with chondrosarcoma ( NCT00949325 ). While awaiting the results of this trial, a study including 10 patients with unresectable chondrosarcoma who were treated with sirolimus and cyclophosphamide showed a disease control rate of 70%. However, the resistance to rapalogs observed in other malignancies suggests that, in chondrosarcoma, a strategy including dual PI3K/mTOR inhibitors such as BEZ235 should be considered for future clinical trials.
Activated Src signaling can also lead to HIF1α expression (see Fig. 3 ) and promote cell survival. Src signaling was shown to be increased in chondrosarcoma, and the tyrosine kinase inhibitor dasatinib showed a decrease in cell proliferation in 7 of 9 cell lines. However, in a phase 2 study, no objective response was obtained with dasatinib single agent (70 mg twice a day as starting dose) in patients with chondrosarcoma (Schuetze CTOS 2010).
Activation of survival pathways can be through stimulation of the receptor tyrosine kinases by IGF-1 or PDGF. IGF-1 pathway activation was shown to be involved in chondrosarcoma proliferation, migration, apoptosis, as well as progression to malignancy. Activation of the PDGF pathway has been shown to be related to worse prognosis in chondrosarcoma. Inhibition with imatinib, however, showed no effect in vitro in 4 chondrosarcoma cell lines, and in a clinical study including 26 patients no objective response was measured. HIF1α expression is suggested to result in increased VEGF expression in chondrosarcoma. Sunitinib and pazopanib are tyrosine kinase inhibitors, targeting multiple kinases including PDGF and VEGF. In combination with proton beam radiation, sunitinib was reported to achieve complete symptomatic relief and durable response in a patient with metastatic clear cell chondrosarcoma. A clinical study with pazopanib is currently recruiting patients with chondrosarcoma ( NCT01330966 ).
Developmental pathways: hedgehog
In osteochondroma, a benign cartilaginous tumor at the surface of bone that can give rise to secondary peripheral chondrosarcoma, mutations in the genes encoding either exostosin-1 ( EXT1 ) or exostosin-2 ( EXT2 ) have been identified. EXT1 and EXT2 are involved in the biosynthesis of heparan sulfate proteoglycans, which are necessary for the diffusion of the morphogen Indian hedgehog (IHH). Recently, osteochondromas were shown to contain a mixture of both EXT mutant as well wildtype tumor cells (with functional EXT), and the latter were shown to be the precursor cells of peripheral chondrosarcoma because peripheral chondrosarcomas have functional EXT, pointing toward a pathogenesis in chondrosarcoma independent of EXT.
IHH is part of a negative feedback loop with parathyroid hormone-related protein (PTHrP), creating a tight balance between proliferation and differentiation (see Fig. 3 ) (for review see Refs. ). Aberrant hedgehog signaling is also found in central chondrosarcoma, despite the absence of EXT mutations. Blocking of the hedgehog pathway with triparanol was shown to be effective, but reports on the effect of cylopamine are conflicting.
A recent randomized phase 2 clinical trial with IPI-926 (saridegib), a potent cyclopamine analogue, for patients with metastatic or locally advanced conventional chondrosarcoma was terminated as the primary end point, progression-free survival, was not met ( NCT01609179 ). A second trial is currently ongoing with vismodegib, a cyclopamine-competitive smoothened inhibitor ( NCT01267955 ). Preliminary results show stable disease in 4 out of 17 patients (Italiano, ASCO 2012). In osteochondroma, primary cilia were found to retain their normal length but lose their orientation contributing to loss of chondrocyte directionality ; 70% to 100% of human enchondromas and chondrosarcomas were found to lack primary cilia. In lft88−/− mice lacking primary cilia, increased hedgehog signaling and enchondroma and chondrosarcoma formation, was observed. As cyclopamine depends on the primary cilia for smoothened accumulation, chondrosarcoma cells lacking primary cilia were unresponsive to cyclopamine treatment. These results support the role for IHH in initiation of chondrosarcoma, and suggest that when inhibiting the hedgehog pathway in chondrosarcoma, targets should be carefully selected.
Developmental pathways: antiapoptosis
The antiapoptotic protein Bcl-2 is under direct regulation of PTH1R and is upregulated in chondrosarcoma (see Fig. 3 ). Moreover, Bcl-xl, another antiapoptotic protein belonging to the Bcl-2 family, was shown to be overexpressed in 18 chondrosarcoma tissues, indicating a specific defense mechanism contributing to chemoresistance in chondrosarcoma. siRNA against Bcl-2, Bcl-xl, and x-linked inhibitor of apoptosis protein showed an enhanced sensitivity to doxorubicin and radiation, and treatment with the BH-3 mimetic ABT-737, was shown to synergistically overcome resistance to doxorubicin and cisplatin. Another antiapoptotic protein, not related to the Bcl-2 family, survivin, was also found to be highly expressed in chondrosarcoma and inhibition experiments in 2 cell lines resulted in overcoming resistance to doxorubicin. These data point toward an effective defense mechanism in which chondrosarcoma cells prevent programmed cell death in response to stress signals such as DNA damage.
Treatment with dulanermin (rhApo2L/TRAIL), a death receptor 4 (DR4) and 5 (DR5) agonist, showed complete remission in 1 patient, and treatment with apomab, a DR5 agonist, showed a 20% reduction in measureable disease in 1 patient with chondrosarcoma, but showed no efficacy in a follow-up phase 2 trial designed to ultimately enroll 90 patients with chondrosarcoma ( NCT00543712 ). These (pre)clinical results combined with this promising result with dulanermin show that restoring the defect in the apoptotic machinery could have strong therapeutic potential in chondrosarcoma. However, because multiple antiapoptotic proteins are upregulated in chondrosarcoma, a multitargeted approach may be more effective, considering that dulanermin, targeting both DR4 and DR5, was more effective than PRO95780, targeting only DR5.
Retinoblastoma signaling
The retinoblastoma protein pRb is a tumor suppressor controlling the cell cycle. In the absence of p16 INK4A , RB-1 is released from E2F transcription factors and cell cycle progression and gene transcription can occur (see Fig. 4 ). Recently Rb was shown to be required for hypertrophic chondrocyte differentiation, and Rb c/c /p107 −/− mice were shown to develop enchondromas, indicating an important role for cell cycle regulation during tumor development.