The improvement in outcome for patients with localized and metastatic Ewing sarcoma since the development of cytotoxic chemotherapy remains one of the most profound advances in oncology and one of the proudest achievements of sarcoma researchers. Identification of molecular targets for new treatments has become an intense area within Ewing sarcoma research. The development of improved preclinical Ewing sarcoma models and advanced molecular techniques will build on knowledge of EWS/FLI1 function, EWS/FLI1 transcription targets, and the other critical driver events in these tumors.
Key points
- •
The improvement in outcome for patients with localized and metastatic Ewing sarcoma since the development of cytotoxic chemotherapy remains one of the most profound advances in oncology.
- •
Vincristine, doxorubicin, and cyclophosphamide alternating with ifosfamide and etoposide (VDC/IE) remains the chemotherapeutic backbone of Ewing sarcoma therapy, and the addition of other cytotoxic agents to this regimen is unlikely to produce significant benefits.
- •
Identification of molecular targets for new treatments has become an intense area within Ewing sarcoma research.
- •
The development of improved preclinical Ewing sarcoma models and advanced molecular techniques, including high-throughput sequencing, will build on knowledge of EWS/FLI1 function, EWS/FLI1 transcription targets, and the other critical driver events in these tumors.
Introduction
The Ewing sarcomas are highly aggressive small round blue cell malignancies of the bone and soft tissue that primarily occur in the second decade of life. The so-called Ewing sarcoma family of tumors includes classical osseous Ewing sarcoma, extraosseous Ewing sarcoma, primitive neuroectodermal tumor, Askin tumor (now thought to be extraosseous Ewing sarcoma of the soft tissues of the chest wall or peripheral lung), and atypical Ewing sarcoma. With variable degrees of neural differentiation, these were initially classified as distinct pathologic entities; however, with advances in molecular profiling, it became clear that these likely derive from the same neuroectodermal cell origin. For example, nearly all Ewing sarcoma family tumors have nonrandom chromosomal translocations involving the Ewing sarcoma breakpoint region 1 gene (EWSR1) on chromosome 22 (described in more detail later). Multimodality therapy, including aggressive neoadjuvant and adjuvant chemotherapy combined with surgery and/or radiation therapy (RT), has greatly improved the long-term survival of patients with localized disease up to greater than 70% at 5 years. Unfortunately, almost 20% of have refractory or recurrent disease and approximatley one-quarter to one-third present with metastatic disease. Despite many attempts at intensifying treatment, survival remains poor in these patients.
Ewing sarcoma remains the second most common primary bone malignancy in the pediatric population, with an annual incidence of almost 3 cases per 1 million people. Reported median age ranges from 12 to 19 ; however, importantly, there is a wide distribution with patients seen at an age of only months up to the 5th and 6th decades of life. Ewing sarcoma is slightly more common in men than women. The incidence is much increased in whites compared with other ethnic backgrounds, which suggests a genetic contribution. Because of its rarity, however, even a modest heritable factor contributing to the risk of developing Ewing sarcoma has not yet been able to be identified.
Clinically, Ewing sarcoma most typically presents with localized symptoms of pain and swelling. In some cases, this is after a prior trauma or muscle strain. Rarely, patients may develop constitutional symptoms, presumably due to cytokine release and inflammation. The most common sites of initial disease include axial and appendicular skeleton with, for example, approximately 20% to 25% originating in the pelvis and approximately 16% to 17% in the femur in some series. Frequent sites of metastatic disease either at diagnosis or recurrence include the lungs and bone.
Adverse prognostic features include location (eg, pelvis ), size of the tumor (>8 cm), high lactate dehydrogenase, time to diagnosis, age (<10–18 years old), and, not surprisingly, evidence of metastatic disease at diagnosis. An important caveat is that many of the negative prognostic factors were identified from somewhat dated patient series and thus are of unknown value with modern chemotherapy. In the authors’ opinion, the most clinically useful negative prognostic factors are those identified in 2 randomized studies using modern chemotherapy regimens (discussed later), which include large tumors, pelvic site, and older age.
In advanced disease, the location of metastases and time to relapse seems to factor into clinical outcome, with improved survival of those with lung versus bone (or bone plus lung) and those with late relapse.
Introduction
The Ewing sarcomas are highly aggressive small round blue cell malignancies of the bone and soft tissue that primarily occur in the second decade of life. The so-called Ewing sarcoma family of tumors includes classical osseous Ewing sarcoma, extraosseous Ewing sarcoma, primitive neuroectodermal tumor, Askin tumor (now thought to be extraosseous Ewing sarcoma of the soft tissues of the chest wall or peripheral lung), and atypical Ewing sarcoma. With variable degrees of neural differentiation, these were initially classified as distinct pathologic entities; however, with advances in molecular profiling, it became clear that these likely derive from the same neuroectodermal cell origin. For example, nearly all Ewing sarcoma family tumors have nonrandom chromosomal translocations involving the Ewing sarcoma breakpoint region 1 gene (EWSR1) on chromosome 22 (described in more detail later). Multimodality therapy, including aggressive neoadjuvant and adjuvant chemotherapy combined with surgery and/or radiation therapy (RT), has greatly improved the long-term survival of patients with localized disease up to greater than 70% at 5 years. Unfortunately, almost 20% of have refractory or recurrent disease and approximatley one-quarter to one-third present with metastatic disease. Despite many attempts at intensifying treatment, survival remains poor in these patients.
Ewing sarcoma remains the second most common primary bone malignancy in the pediatric population, with an annual incidence of almost 3 cases per 1 million people. Reported median age ranges from 12 to 19 ; however, importantly, there is a wide distribution with patients seen at an age of only months up to the 5th and 6th decades of life. Ewing sarcoma is slightly more common in men than women. The incidence is much increased in whites compared with other ethnic backgrounds, which suggests a genetic contribution. Because of its rarity, however, even a modest heritable factor contributing to the risk of developing Ewing sarcoma has not yet been able to be identified.
Clinically, Ewing sarcoma most typically presents with localized symptoms of pain and swelling. In some cases, this is after a prior trauma or muscle strain. Rarely, patients may develop constitutional symptoms, presumably due to cytokine release and inflammation. The most common sites of initial disease include axial and appendicular skeleton with, for example, approximately 20% to 25% originating in the pelvis and approximately 16% to 17% in the femur in some series. Frequent sites of metastatic disease either at diagnosis or recurrence include the lungs and bone.
Adverse prognostic features include location (eg, pelvis ), size of the tumor (>8 cm), high lactate dehydrogenase, time to diagnosis, age (<10–18 years old), and, not surprisingly, evidence of metastatic disease at diagnosis. An important caveat is that many of the negative prognostic factors were identified from somewhat dated patient series and thus are of unknown value with modern chemotherapy. In the authors’ opinion, the most clinically useful negative prognostic factors are those identified in 2 randomized studies using modern chemotherapy regimens (discussed later), which include large tumors, pelvic site, and older age.
In advanced disease, the location of metastases and time to relapse seems to factor into clinical outcome, with improved survival of those with lung versus bone (or bone plus lung) and those with late relapse.
Molecular biology
Within the sarcomas, there are subsets with defined genetic alterations, including those with translocations involving members of the ten-eleven translocation (TET) family of RNA-binding proteins (including translocated in liposarcoma/fused in sarcoma [TLS/FUS], EWS, and TATA-binding protein–associated factor 15 [TAF15], reviewed by Tan and Manley and by Jain and colleagues ). Examples include the Ewing family of tumors, desmoplastic small round cell tumor, clear cell sarcoma, angiomatoid fibrous histiocytoma, extraskeletal myxoid chondrosarcoma, and myxoid/round cell liposarcoma.
The TET gene products have multiple putative cellular functions, including roles in regulating transcription, RNA splicing, and intracellular signaling. In all TET family proteins, there is an RNA-binding domain localized to the C-terminus and an activation/regulatory domain is within the N-terminus. These sarcomas are characterized by aberrant gene chimerisms, which bring together the TET amino terminal activation domain with the DNA-binding domain of a partner transcription factor. Thus, it is thought that this fusion protein may drive altered gene expression, cell differentiation, and ultimately tumor growth. The TET member and transcription factor member can be substituted within their subgroups while still resulting in similar histologic phenotypes. For example, in most (approximately 85%–90%) Ewing sarcoma tumors, the EWS gene 22 is aberrantly juxtaposed to the ETS transcription factor FLI1 on chromosome 11: t(11;22)(q24;q12). Other ETS translocation partners with EWS, however, include ERG1 (10%), ETV1, ETV4 (also known as E1AF), and FEV. Additionally, the TET family member TLS/FUS is rarely seen to translocate to ERG1 and FEV in a phenotype indistinguishable from those with EWS.
Initial evaluation and treatment
Given the infrequency of this disease and the intricacies of clinical management, it is highly recommended that Ewing sarcoma patients undergo initial evaluation and treatment planning at a high-volume sarcoma treatment center. Histologic and molecular diagnosis should be confirmed by an expert pathologist. Ewing sarcomas are small blue round cell tumors ( Fig. 1 ) typically positive for CD99 staining with variable degrees of neuronal differentiation. EWS rearrangement is either confirmed with fluorescence in situ hybridization (FISH) or by polymerase chain reaction. The FISH probe does not identify the translocation partner and (as discussed previously) EWS translocations can occur in other sarcomas. Moreover, in unpublished personal observation of a single patient under the authors’ care, the EWS FISH probe was negative but molecular testing with DNA sequencing did identify EWS/FLI1 (Choy E, unpublished, 2012). Thus, if clinical suspicion is high, sequencing or polymerase chain reaction–based studies could be obtained.
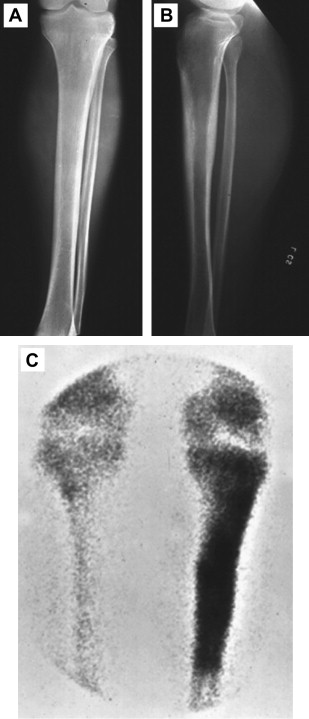
The pretreatment evaluation should include full staging studies. Traditional plain films show a moth-eaten area of bone, revealing a confluence of finely destructive lesions that coalesce over time. Standard radiographs ( Fig. 2 ) are now often supplanted by diagnostic CT scans in upfront imaging, which can assess the degree of bone cortex that is compromised by the tumor and predict the risk of developing pathologic fractures. MRI remains the gold standard for radiographic characterization of Ewing sarcoma because it can determine the relationship of tumor with critical anatomic structures, vessels, nerves, and surgical/fascial planes. All patients should also receive chest CT and technetium Tc 99m bone scanning or positron emission tomography scanning to identify metastases to the lungs and bones, respectively. Some practitioners perform bone marrow aspirates or MRI of the spine and pelvis to rule out occult marrow involvement. It is now the standard of care, however, to treat all chemotherapy-eligible patients with up to 1 year of systemic therapies regardless of proof of metastasis (discussed later), and as such, the bone marrow aspirate has recently played a lesser role in determining the clinical management of patients.
Localized Ewing Sarcoma
Despite up to 75% of patients having clinically and radiographic evidence of only localized tumors, it is known that without systemic chemotherapy most of these patients go on to develop overt metastatic disease. This has led to the hypothesis and clinical assumption in practice that patients with radiographically confirmed localized disease (with or without bone marrow biopsy) actually have micrometastatic Ewing sarcoma that is below the detection limits of staging techniques.
The current standard of care in the United States involves 17 cycles of chemotherapy using VDC/IE (vincrisine, doxorubicin, cyclophosphamide/ifosfamide, etoposide). Typically, patients receive 4 to 6 cycles of neoadjuvant therapy followed by definitive local control (eg, surgery, RT, or surgery and RT) with continued chemotherapy for up to nearly a year, as long as there is evidence of sustained tumor response. This neoadjuvant/adjuvant strategy allows for rapid systemic and local control, the improvement of surgical options or radiation fields, and the assessment of tumor response on the surgical specimen. This intense, lengthy, and somewhat unusual regimen has been developed over several decades through several cooperative studies. A brief history is as follows.
The Intergroup Ewing’s Sarcoma Study (IESS)-I recruited 342 children with localized tumors from 1973 to 1978. Patients were randomized to radiation to the primary lesion plus (1) vincristine/doxorubicin/cyclophosphamide/dactinomycin (VDCA), (2) vincristine/cyclophosphamide/dactinomycin, or (3) vincristine/cyclophosphamide/dactinomycin plus bilateral prophylactic pulmonary RT. The 5-year relapse-free survival was superior for VDCA at 60% compared with 24% and 44% for treatments 2 and 3, respectively. Overall survival was similarly improved at 5 years: 65% alive in treatment group 1 and only 28% and 53% in treatments groups 2 and 3, respectively. Thus, doxorubicin was established as a key addition to the regimen.
IESS-II, the subsequent intergroup study, was designed to determine if increasing the dose intensity could improve outcome in osteosarcoma and Ewing sarcoma patients. This study randomized 214 nonmetastatic Ewing sarcoma patients to VDCA with a high-dose, intermittent dosing strategy compared with a lower weekly dosing schedule. Specifically, high-dose cyclophosphamide given every 3 weeks was compared with lower-dose weekly treatments. Additionally, doxorubicin was given every 6 weeks for 9 months compared with alternating doses with dactinomycin. The higher dose–intensity patients showed a 5-year disease-free survival of 68% compared with 48% in the less-intense treatment arm. Overall survival was 77% versus 63%. At this point, evidence suggested that upfront anthracycline and higher-dose alkylators were necessary to improve outcome.
There are several subsequent studies that demonstrated the benefit of adding ifosfamide and etoposide (IE) to the core regimen of VDC (the rationale of which was based on activity observed in the relapsed setting). Two studies from the Rizzoli Orthopedic Institute in Bologna, Italy, REN-2 and REN-3, combined IE with VDCA. In REN-2, IE was combined with VDCA in a maintenance strategy. In a single treatment arm, 82 patients received neoadjuvant VDC followed by alternating cycles incorporating IE. At 5 years, disease-free survival was 54% and overall survival 59%. The follow-up study, REN-3, treated 157 patients with nonmetastatic Ewing sarcoma with neoadjuvant VDC and vincristine/ifosfamide/dactinomycin followed by cycles of VDC, vincristine/ifosfamide/dactinomycin, IE, and vincristine/cytoxan/dactinomycin. This complicated regimen first brought ifosfamide into the neoadjuvant stage of treatment and had an improved event-free survival of 71% and overall survival of 76.5% compared with historical controls.
The Children’s Oncology Group (including the Pediatric Oncology Group [POG] and Children’s Cancer Group [CCG]) designed a randomized trial of 518 patients with localized or metastatic Ewing sarcoma, ages 30 years and younger, where patients received 49 weeks (17 cycles) of VDC alone or alternating cycles of VDC with IE. Dactinomycin replaced doxorubicin after patients received the 375 mg/m 2 of doxorubicin. Local control was planned at week 12 via RT or surgery or both. In the 398 patients without metastatic disease, the 5-year event-free survival was 69% for the VDC/IE arm versus 54% in the VDC arm ( P = .005). Overall survival was similarly improved, at 72% and 61% ( P = .01), respectively.
The POG/CCG attempted to build on to this VDC/IE regimen by compressing the same total doses of ifosfamide and cyclophosphamide into 11 high-dose cycles every 3 weeks compared with 17 standard doses every 3 weeks ; 487 participants were randomized to either arm. Unfortunately, there was no difference in event-free survival and, not surprisingly, toxicity was higher in the dose-intense regimen.
Given the disappointing results of dose intensification, the Children’s Oncology Group designed another large randomized study to evaluate the efficacy of increasing dose frequency rather than intensity ; 568 patients without evidence of metastatic Ewing sarcoma were randomized to either the standard or dose-dense arm. In the standard arm, patients received VDC/IE every 3 weeks for 2 cycles (4 treatments) followed by local control and then VDC/IE every 3 weeks for 5 doses (10 more treatments). Doxorubicin was limited to a cumulative dose of 375 mg/m 2 and was dropped from subsequent cycles (ie, no dactinomycin was given in this trial). In the experimental arm, patients received VDC/IE every 2 weeks with growth factor support for 3 cycles (6 treatments) followed by local control and then VDC/IE every 2 weeks with growth factor for 4 more cycles (8 more treatments). Again doxorubicin was limited to 375 mg/m 2 . The event-free survival at 5 years was 73% for the 14-day regimen and 65% for the 21-day regimen ( P = .048). Overall survival was not significantly improved at 83% and 77% ( P = .056), respectively, however. The toxicity of the regimens was similar between the arms. Subgroup analysis identified worse outcome in with patients with pelvic primaries and/or age over 18 years old. There were, however, only 67 patients over 18 years old in the study so it is impossible to extrapolate whether the intensified regimen improved outcomes in these high-risk subgroups.
Regarding the primary lesion, there is no current level 1 data on a head-to-head comparison of RT versus surgery versus both RT and surgery in the local management of Ewing sarcoma. Retrospective studies are inherently difficult to interpret due to selection bias. Practice patterns are clinician and center dependent. Many factors are weighed, including the location of disease, the ability to achieve an acceptable surgical outcome (eg, margins, function, and morbidity), and the risks of RT, such as a secondary malignancy and the effects on bone function and growth.
Thus, taken together, standard of care at the authors’ institution is VDC/IE given either as dose dense (every 2 weeks) for 14 cycles or every 3 weeks for 17 cycles. There is variable practice between physicians of substituting dactinomycin for doxorubicin after 375 mg/m 2 versus dropping it all together. Although tempting, statistically, the first VDC/IE randomized study (which included dactinomycin) should not be compared with the dose-dense strategy (which omitted dactinomycin) despite the similar event-free and overall survival rates. Because the authors’ practice treats primarily adult patients, the decision to attempt a dose-dense strategy is made on a patient-to-patient basis, depending on comorbidities, age, and tolerance of the regimen. Finally, regarding imaging during treatment, there is much variability between studies (discussed previously) and there are no formal United States guidelines. The authors believe, at minimum, that the generally accepted time points for staging include at diagnosis, before definitive tumor therapy (ie, surgery or RT or both), and at completion of treatment. Imaging should include targeting the primary site plus a chest CT; some practitioners include positron emission tomography or bone scans in these later time points. The authors typically repeat primary lesion imaging and a chest CT every 3 months after the completion of chemotherapy. During surveillance after therapy, the US National Comprehensive Cancer Network guidelines suggest physical examination, laboratory tests, and primary site plus chest imaging every 2 to 3 months for 2 years. Thereafter, intervals can be increased up to annually after 5 years. Special attention should be made to long-term complications at these late visits, including secondary RT or chemotherapy-associated malignancies and cardiovascular disease.
Metastatic and Recurrent Ewing Sarcoma
For the approximately 20% of patients with grossly metastatic Ewing sarcoma at presentation, survival remains poor. For example, in the studies discussed previously, 5-year disease-free survival was disappointing and ranged from 9% to 30%. Surveillance, Epidemiology and End Results data suggest that even in the metastatic setting there are some patients that can achieve long-term survival.
Published studies, albeit with small numbers, have explored similar systemic chemotherapy regimens (described previously) for localized disease, including doxorubicin/cylophosphamide, VDCA, and VDCA plus fluorouracil and VDC/IE. Probably the most important of these is the POG/CCG study, where 120 patients with metastatic disease were randomized to either VDC or VDC/IE. The 5-year event-free survival was 22% in either treatment arm and overall survival was 34% to 35%. Thus, there was no statistically significant impact on mean overall survival by adding IE to the VDCA backbone. Similar data were seen in single-arm feasibility studies.
Attempts at dose intensification with and without autologous or allogeneic stem cell rescue have been somewhat disappointing. The most recent, and largest, of these was the Euro-EWING 99 trial, which recruited 281 patients with metastatic Ewing sarcoma from 1999 to 2005. Treatment included 6 cycles of vincristine/ifosfamide/doxorubicin/etoposide followed by 1 cycle of vincristine/dactinomycin/ifosfamide before local therapy. The patients were then conditioned with high-dose chemotherapy with busulfan and melphalan with autologous stem cell rescue. Safety was comparable to other regimens during induction and autologous stem cell rescue, with 1 patient dying during induction and 2 within the first 100 days of transplant. The event-free and overall survival rates were 27% and 34%, respectively. These results are comparable to other small reported studies. Thus, although there are some patients who can achieve a durable response, the authors believe that these studies should be reserved for the setting of a clinical trial.
At first diagnosis at the authors’ institution, metastatic patients are typically treated with standard VDC/IE or VDC alone with the hope that some of these patients can be salvaged if they achieve responses. The selection of the regimen is based on individual patient characteristics, including symptoms, age, and limited pulmonary metastases. Other regimens that could be considered include vincristine/doxorubicin/ifosfamide and vincristine/ifosfamide/doxorubicin/etoposide. RT is used to treat the primary and other sites based on symptoms alone. Surgical resection may be considered depending on response and the ability to remove all sites of disease (eg, oligometastatic disease in the lung).
Outcomes in the progressive and recurrent setting also remain poor where there is no standard of care and complete responses are rare. In a single-center case series of 114 patients, only 12.3% remained alive at a median follow-up of 61 months. Common regimens include ifosfamide/etoposide (if not given at first treatment), ifosfamide/carboplatin/etoposide, docetaxal/gemcitabine, topotecan/cyclophosphamide, and temozolomide/irinotecan, The authors typically favor the latter 2 regimens, which combine topoisomerase I inhibitors (topotecan and irinotecan) with alkylators (cyclophosphamide and temozolomide), with reported objective response rates ranging from approximately 30% to 60%. With initial metastatic treatment or at relapse, however, the authors’ bias is that early access to clinical trials should be offered to patients where available because, ultimately, it is clear that the limit of chemotherapy in the metastatic setting has been reached.
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


