From 1976 to 2010, only 2 medications were approved for treating metastatic melanoma. Between 2011 and 2013, 4 agents were approved and other therapies have shown great promise in clinical trials. Fundamental discoveries, such as the identification of oncogenic mutations in most melanomas, the elucidation of the molecular signaling resulting from these mutations, and the revelation that several cell surface molecules serve as regulators of immune activation, have been instrumental in this progress. This article summarizes the molecular pathogenesis of melanoma, describes the current efforts to target oncogene-driven signaling, and presents the rationale for combining immune and molecular targeting.
Key points
- •
Melanomagenesis is a complex process that involves carcinogenic exposure (eg, ultraviolet B radiation) and genetic predisposition (MCR1 polymorphisms).
- •
Microphthalmia-associated transcription factor, the “master regulator of melanocyte development,” functions as an oncogene when dysregulated, leading to the upregulation of cell cycle progression and favorable metabolic changes, and the inhibition of apoptosis.
- •
Mutations of oncogenes (BRAF, NRAS, CKIT, GNAQ, GNA11) and a tumor suppressor gene (NF1) are present in most melanomas, although the rate of each type of mutation varies according to anatomic site subset (eg, mucosal vs cutaneous vs uveal).
- •
The mitogen-activated protein kinase (MAPK) pathway is upregulated in nearly all melanomas and is susceptible to small molecule inhibition.
- •
The interplay among the MAPK pathway, tumor antigen expression, and immune infiltration predicts therapeutic synergy with combined molecular and immune targeting.
Introduction
Over the past 3 decades, several breakthroughs have greatly expanded what is known about melanocyte biology, relevant oncogenic mutations and deletions and amplifications in melanoma, the influence of molecular signaling pathways on melanomagenesis, and the interaction of aberrant signaling pathways with host immune elements. This increased understanding has led to a remarkable number of improvements in the diagnosis, classification, and treatment of this disease. This advancement is an amazing achievement with great relevance, because the number of new cases of and deaths from melanoma continues to increase. A hallmark of this recent success is the regulatory approval of 4 therapeutic agents over the past 3 years, with at least another 6 promising agents that have just entered or completed phase III clinical trials. Still, most patients diagnosed with metastatic melanoma will die of their disease within a few years of diagnosis. To achieve the goal of successfully treating metastatic melanoma for nearly all afflicted, continued breakthroughs will be required to provide clinicians with diagnostic tools to identify subsets of patients most likely to benefit from a specific line of therapy, and improved treatment strategies for these identified subsets. This article reviews the relevant discoveries regarding melanocyte and melanoma biology that have been or are beginning to be translated into transformative therapies.
Introduction
Over the past 3 decades, several breakthroughs have greatly expanded what is known about melanocyte biology, relevant oncogenic mutations and deletions and amplifications in melanoma, the influence of molecular signaling pathways on melanomagenesis, and the interaction of aberrant signaling pathways with host immune elements. This increased understanding has led to a remarkable number of improvements in the diagnosis, classification, and treatment of this disease. This advancement is an amazing achievement with great relevance, because the number of new cases of and deaths from melanoma continues to increase. A hallmark of this recent success is the regulatory approval of 4 therapeutic agents over the past 3 years, with at least another 6 promising agents that have just entered or completed phase III clinical trials. Still, most patients diagnosed with metastatic melanoma will die of their disease within a few years of diagnosis. To achieve the goal of successfully treating metastatic melanoma for nearly all afflicted, continued breakthroughs will be required to provide clinicians with diagnostic tools to identify subsets of patients most likely to benefit from a specific line of therapy, and improved treatment strategies for these identified subsets. This article reviews the relevant discoveries regarding melanocyte and melanoma biology that have been or are beginning to be translated into transformative therapies.
Melanoma development
Melanocyte Formation in Development
Melanocytes are neural crest–derived cells that develop as a branch of alternative differentiation programs that include the closely related lineages of sympathetic neurons, Schwann cells, or melanocytes. In addition to residing in the basal epidermis and in hair follicles, other melanocyte populations can be found along mucosal surfaces and in the meninges, choroidal layer of the eye, and stria vascularis within the cochlea. The pigments produced by melanocytes are composed of numerous chemical species that have been broadly classified as red/blond pigments (pheomelanin) and brown/black pigments (eumelanin). Although brown/black pigment has a measureable (albeit modest) ultraviolet protective capability, pheomelanin has been associated with increased reactive oxygen species in the skin.
Two forms of skin pigmentation exist: constitutive and adaptive. The constitutive or basal skin pigment level is associated with the type of pigment synthesized and the maturation process of the melanin-containing vesicles (called melanosomes). People of varying constitutive pigmentation are thought to have a constant number of melanocytes but variations in relative pigment production per cell. The adaptive pigmentation response typically reflects melanin synthesis triggered by ultraviolet radiation. This pigment has been shown to be initiated by ultraviolet-induced DNA damage in overlying epidermal melanocytes followed by p53 stabilization and transcriptional activation of the proopiomelanocortin (POMC) gene. POMC is posttranslationally cleaved into various small peptides, one of which is the melanocyte-stimulating hormone (MSH) that is secreted and stimulates its receptor (melanocortin receptor 1 [MC1R]) on underlying melanocytes. Activation of MC1R by MSH peptide results in cyclic adenosine monophosphate induction within melanocytes, followed by stimulation of the gene encoding a transcription factor called microphthalmia-associated transcription factor (MITF), which activates expression of all known pigment-producing enzymes and nearly all of the machinery required for the packaging, maturation, and secretion of pigment-laden melanosomes. Nonfunctional polymorphic variants of MC1R are frequently responsible for the red hair/fair skin/freckling phenotype in numerous species, including humans.
Role of Ultraviolet Radiation
Ultraviolet radiation is deeply implicated in the formation of common forms of cutaneous melanoma in man. Ultraviolet wavelengths residing within the ultraviolet B portion of the spectrum produce stereotypical nucleotide adducts known as cyclobutane pyrimidine dimers , which in turn result in formation of pyrimidine dimer mutations, in which a cytosine located in a dipyrimidine sequence becomes mutated to thymidine. These “ultraviolet signature mutations” are easily recognizable within irradiated DNA. Exomic deep sequencing of human melanomas has been performed recently, and clearly reveals a striking abundance of these ultraviolet-derived genomic mutations within melanomas. Still, features of melanoma suggest contributions in addition to ultraviolet radiation, such as red/blond pigment, that may provide new opportunities to improve prevention strategies.
Role of MITF
Microphthalmia-associated transcription factor is a transcriptional regulator of the pigment pathway in melanocytes. However, MITF deficiency affects melanocytes more profoundly than purely affecting pigment synthesis. MITF deficiency produces Waardenburg syndrome type 2A in man and complete absence of the melanocyte lineage in numerous species. This vital role for MITF in melanocyte development has led to its being dubbed a “master regulator of melanocyte development.” In contrast to consequences of MITF deficiency, more recent studies showed that genomic amplification of MITF, a recurrent activating germline point mutation, activates melanoma oncogenes in man. The recurrent point mutation in MITF was identified in cases of familial melanoma, and resulted in disruption of a SUMO-modification consensus sequence, thereby modestly increasing transcriptional activity of MITF protein. The means through which MITF dysregulation contributes to melanocytic transformation to malignancy is still being determined. It is unlikely that MITF’s oncogenic activity is related to its production of eumelanin (dark eumelanin pigment is likely to be protective against melanoma and nonmelanoma skin cancers). However, MITF has also been seen to directly regulate expression of antiapoptotic genes (BCL2, BCL2A1), the cell cycle regulator CDK2, the metabolic regulator PGC1α, and numerous others. Because of the role of MITF as a lineage survival factor, its role as an active regulator of melanoma proliferation/survival is likely to occur in most melanomas regardless of whether it is also an amplified or mutated oncogene. Its direct phosphorylation by mitogen activated protein kinase (MAPK) and subsequent ubiquitin-dependent proteolysis link MITF activity to the common melanoma oncogenes BRAF and NRAS.
Molecular signaling in melanoma
MAPK Signaling
The MAPK signaling pathway is critical to the pathobiology of several malignancies, including melanoma. Canonical signaling ( Fig. 1 ) through the pathway begins with ligand–receptor, either receptor tyrosine kinase (RTK) or G-protein–coupled receptor (GPCR) activation, engagement at the cell surface that ultimately triggers recruitment and activation of 1 of 3 (H-, K-, and N-) RAS isoforms. Once activated, RAS binds to the RAS-binding domain on 1 of 3 (A-, B-, C-) RAF isoforms, triggering RAF homodimerization or heterodimerization and activation of its serine-threonine kinase domain. Phosphorylation of MAPK kinase (MEK) by RAF leads its activation, which then leads to the activation of its only known substrate, MAPK otherwise known as the extracellular signal-regulated kinase (ERK). The activation of ERK then leads to a host of downstream events that promote cell cycle progression and cell survival.
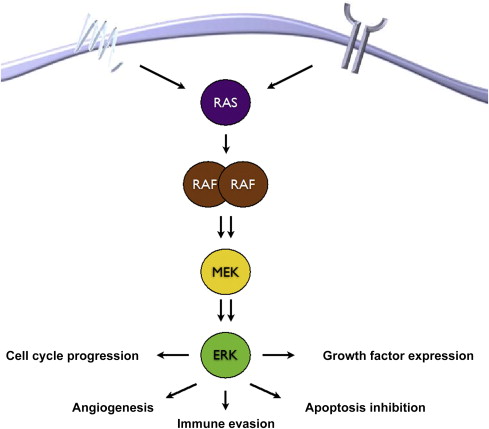
Deregulation of cell cycle progression is a critical consequence of MAPK activation in melanoma, and can occur through genomic mutations and amplifications (discussed later) or through hyperactivation of ERK. ERK phosphorylation and cyclin D1 expression, measured by immunohistochemistry staining, have been correlated in the analysis of primary melanomas. Additionally, in uveal and acral melanoma, increased MAPK signaling has been shown to be associated with cyclin D expression in the absence of oncogenic mutations of either BRAF or NRAS. In the setting of oncogenic BRAF mutations, the cyclin-dependent kinase (CDK) inhibitor p27 KIP1 is downregulated and cyclin D1, is constitutively activated, both in an ERK-dependent manner, leading to enhanced proliferation. In the setting of oncogenic NRAS mutations, cyclin D1 also seems to be regulated through ERK phosphorylation and plays an important role in driving proliferation.
MAPK signaling also leads to the inhibition of apoptosis in melanoma cells. This process likely occurs through several mechanisms, although most of its effects seem to be in the regulation of various proapoptotic and antiapoptotic B-cell lymphoma 2 (BCL2) family members, including BIM, BAD, BMF, MCL-1, BAX, NOXA, and PUMA. The preponderance of evidence suggests that the most critical effects of MAPK hyperactivation, particularly via oncogenic BRAF, on apoptosis are exerted through inhibition/downregulation of the proapoptotic BH3-protein BIM. This effect may occur through several mechanisms, including decreased RNA expression, increased antiapoptotic BCL2 family member expression, or alterations in the BIM splicing machinery.
Phosphoinositol-3-Kinase Signaling
The phosphoinositol-3-kinase (PI3K) pathway is the second major signaling pathway involved in melanoma pathogenesis. Canonical activation of the PI3K pathway begins, much like the MAPK pathway activation, with cell surface receptor–ligand engagement, thereby triggering conversion of PIP2 to PIP3 that then binds to and activates Akt. Activated Akt leads to several cellular processes that promote cell proliferation, cell survival, and angiogenesis. In melanoma, the PI3K pathway may be activated via several mechanisms, including NRAS activation, either through mutation or loss of neurofibromatosis 1 (NF1) function, which can activate PI3K in a receptor-independent mechanism, loss of function of the PI3K regulator, phosphatase and tensin homologue (PTEN), or oncogenic mutation of Akt3. One of the key downstream events of Akt activation is the activation of the mammalian target of rapamycin complexes 1 and 2 (mTORC1 and mTORC 2) through the inhibition of the tuberous sclerosis complex 1 and 2 (TSC1/TSC2), a tumor suppressor protein that regulates mTOR. mTORC1 and mTORC2 activation then lead to several cellular events, often hallmarked by the phosphorylation of the ribosomal protein S6 (phosphor-S6), that lead to proliferation, angiogenesis, and resistance to apoptosis.
TSC1/TSC2 expression is regulated by several molecules, including AMP kinase 1 (AMPK1), which is turn is activated by liver kinase B1 (LKB1). This pathway seems to be important in melanoma, particularly BRAF-mutant melanoma, because oncogenic BRAF is associated with downregulation of LKB1, and LKB1 expression is increased in the setting of BRAF inhibitor therapy. This mechanism serves as a critical connection between the MAPK and PI3K signaling pathways that may have prognostic and therapeutic implications. Specifically, a recent analysis of patients treated with BRAF inhibitors showed that outcome of therapy was significantly improved when on-treatment phospho-sS6 was absent compared with when it was preserved. It is conceivable that incorporating an “early look” at phospho-S6 expression in patients treated with BRAF inhibitors could help clinicians switch to a broader molecular-targeted regimen that incorporates PI3K pathway inhibition.
Oncogenic Mutations
The transformation of melanocytes into melanoma cells is a complex process that involves the acquisition of multiple molecular aberrations. These aberrations include genetic mutations and amplifications of well-described oncogenes that lead to hyperactivation of signal transduction pathways that promote cell growth, angiogenesis, survival, and immune evasion. The pattern of mutations varies depending on where the melanoma arises ( Fig. 2 ).
BRAF
Oncogenic mutations in the kinase domain of the serine/threonine kinase BRAF are present in nearly 50% of melanomas. These mutations most commonly involve the valine at the 600 position (V600), whereby this is exchanged for a glutamic acid (V600E) or an arginine (V600K). Other mutations have also been described in BRAF, but the V600 mutations dominate, representing most (>95%) of the BRAF mutations identified in melanoma, and are associated with constitutive activation of the MAPK pathway. In the setting of oncogenic BRAF mutations, RAS-RAF engagement and RAF dimerization is not required for RAF-induced activation of MEK and ERK.
BRAF V600E mutations are the most common BRAF mutations and occur more commonly in women, younger patients, and non–sun exposed areas. In contrast, BRAF V600K mutations occur more frequently in older patients and men in sun-exposed areas. These mutations seem to be more frequent in geographic locations with more intense sun exposure, such as Australia and Texas, and less common in the Northeast. Both mutations lead to a constitutively activated kinase that signals in monomeric form and is susceptible to inhibition with small molecules. Three such agents, vemurafenib, dabrafenib, and LGX818 have shown significant clinical activity, with both vemurafenib and dabrafenib having achieved regulatory approval for the treatment of patients with BRAF V600E/K -mutant melanoma.
NRAS/NF1
The N -isoform of RAS is mutated in 15% to 25% of patients with melanoma, and seems to be represented in similar frequencies in all anatomic subsets of the disease. NRAS mutations seem to be mutually exclusive to BRAF mutations in most cases (dual positivity is seen at an incidence of <0.5%), and direct oncogenic signaling through the MAPK and PI3K pathways. In addition, the tumor suppressor gene neurofibromatosis 1 (NF1), a potent regulator of NRAS, is dysfunctional in another 10% to 15% of cases, which leads to upregulation of NRAS signaling. These recently identified loss-of-function mutations of NF1 are probably associated with similar activation of the MAPK and PI3K pathways as are activating NRAS mutations, and may be susceptible to the same treatment approaches.
CKIT
The tyrosine kinase receptor CKIT provides growth-pathway activation after interaction with and activation by its ligand stem cell factor. This pathway is critical in several normal cellular processes, including melanocyte pigment production. However, mutations and genomic amplifications in the kinase or transmembrane domains lead to constitutive activation of the receptor and are associated with several malignancies, including melanoma and gastrointestinal stromal tumor (GIST). In melanoma, mutations are typically seen in the transmembrane domains coded in exons 11 and 13, are more commonly found in acral/lentiginous and mucosal melanomas, and may be sensitive to small molecule inhibition with CKIT inhibitors, such as imatinib.
GNAQ/GNA11
G-protein coupled receptors are 7-transmembrane domain receptors that transduce signals from several types of ligands, including hormones, growth factors, cytokines, and chemokines. Mutations in the 1 of 2 G-proteins, GNAQ and GNA11, have been identified in most patients with uveal melanoma. In this setting, these constitutively activated proteins lead to the activation of protein kinase C, which triggers MAPK pathway activity through MEK phosphorylation, and renders these cells susceptible to MEK and PKC inhibitor therapy.
Other mutations
Although BRAF, NRAS, CKIT, GNAQ/GNA11, and NF1 mutations are primary mutations that are typically mutually exclusive of each other, several other mutations or genetic aberrations affect the oncogenic phenotype in melanoma.
Cell cycle regulation
Dysregulation of the cell cycle at the genomic level is common in melanoma. The most commonly aberrant cell cycle–regulating gene in melanoma is CDNK2A, which codes for p16 INK4A , a CDK inhibitor, and is either deleted or mutated in at least 50% to 60% of tumors. Additionally, loss of p16 can occur through promoter methylation in up to 10% to 20% of cases; thus up to 60% to 80% of patients may have disruption of p16 INK4A expression or function. These aberrations lead to deregulation and overexpression of CDK4, which contributes to oncogenesis in the setting of other oncogenic drivers, such as NRAS and BRAF mutations.
Most melanomas are sporadic; however, germline mutations of CDNK2A are the most commonly inherited mutation associated with hereditary melanoma. Affected individuals have an approximately two-thirds lifetime risk of developing melanoma. A second, hereditable form of melanoma involves germline-activating mutations of CDK4. Although these are not as common as CDNK2A mutations, they are similarly associated with multiple nevi, early-onset melanoma, and the development of multiple primary melanomas.
Mutations in the tumor suppressor p53 are common in human malignancies, although they are infrequently seen in patients with melanoma. However, amplification and overexpression of MDM2, a protein that ubiquitinates p53 therapy, leading to its jettisoning from the nucleus and proteasomal degradation, is present in more than 10% and 50% of patients with melanoma, respectively.
PTEN loss
PTEN is a tumor suppressor gene/protein and the major regulator of PI3K activation. Mutations leading to a truncated and/or nonfunctional protein and deletion of PTEN (so-called PTEN loss) are seen in approximately 25% of patients with melanoma. These mutations are most commonly seen concomitant with BRAF mutations, although they rarely occur in the setting of NRAS mutations or BRAF and NRAS wild-type melanomas. In the context of oncogenic BRAF mutations, PTEN loss is associated with PI3K pathway signaling and reduced effectiveness of BRAF inhibitors. Recent data suggest that PTEN loss is specifically associated with PI3Kβ signaling, and the first PI3Kβ inhibitors currently are being tested in a clinical trial ( ClinicalTrials.gov identifier: NCT01673737 ). Dual targeting of the MAPK pathway and PI3Kβ is expected to be explored early in the development of these agents.
Oncogene-directed treatments
The identification of targetable mutations in melanoma, as with other malignancies such as chronic myelogenous leukemia, non–small cell lung cancer, and GIST, has led to a robust clinical development effort of small molecule inhibitors for the treatment of melanoma. As depicted in Fig. 3 , several potential types of agents are being tested in patients with molecularly defined subtypes of melanoma. The most success to date has involved inhibitors of BRAF and MEK in BRAF-mutant melanoma and, to a lesser extent, MEK and KIT inhibitors in NRAS-mutant and CKIT-mutant melanoma, respectively. Although the clinical data from these studies is addressed elsewhere in this issue, 2 key principles of targeted therapy development in melanoma are addressed here.
The first principle of targeted therapy drug development is to use effective inhibitors of the target. With the identification of oncogenic BRAF mutations in almost all patients, inhibitors of RAF generally and BRAF specifically were brought quickly to the clinic. The first 2 RAF inhibitors to make it to early-phase clinical trials were sorafenib and RAF265. Both are pan-RAF inhibitors, meaning they inhibit BRAF and CRAF, and the mutant forms of BRAF. In preclinical studies, these agents had broad activity against BRAF mutant and wild-type cell lines. Although some initial enthusiasm was shown for sorafenib in combination with chemotherapy, it was proven ineffective as a single agent and in combination with chemotherapy in numerous phase I, II, and III trials. Additionally, RAF265 has been shown to be similarly ineffective. With each agent, both on- and off-target toxicity limited the drug exposures predicted for efficient RAF inhibition, and likely explains the relative ineffectiveness of these agents compared with the more selective BRAF inhibitors that either have been approved or are in clinical development. The first of these to demonstrate effectiveness in patients with melanoma was vemurafenib, a more selective RAF inhibitor that has nearly 10-fold greater inhibition of mutant BRAF versus wild-type BRAF. In dose-escalation of this agent, drug exposure was lower than predicted and not dose-dependent; treatment with this formulation was associated with minimal activity. With reformulation, drug exposure was dose-dependent, and most patients with BRAF mutations experienced disease regression. Clinical activity was confirmed in phase II and III trials, leading to its approval by the U.S. Food and Drug Administration (FDA) in 2011.
The second principle of effective drug development is careful patient selection. The development of the KIT inhibitor imatinib for the treatment of melanoma provides an excellent example of this principle. In fact, the clinical development of imatinib in melanoma was nearly permanently derailed by 2 phase II trials performed in patients with stage IV melanoma. The genesis of these trials was that KIT expression, measured through immunohistochemistry testing, was seen in most patients with melanoma; although no responses were seen in these trials of 18 and 29 patients, survival was poor and toxicity high, and although immunohistochemistry expression was seen in only greater than 50% of samples that were available, this did not seem to correlate to outcome. Since the completion of these trials, mutations and amplifications of CKIT have been identified (as described earlier), and in 3 phase II trials enriched with patients with either CKIT mutation or amplification, response rates have ranged from 17% to 29% in all patients and 30% to 54% in patients with mutation or amplification. This example highlights the concept that an inhibitor will only be effective when given to the patients most likely to benefit. As targeted therapy development continues in melanoma, appropriate definition of molecular subsets remains one of the most critical factors to get right.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


