59 Tumors of Bone and Soft Tissue
Radiographic Features
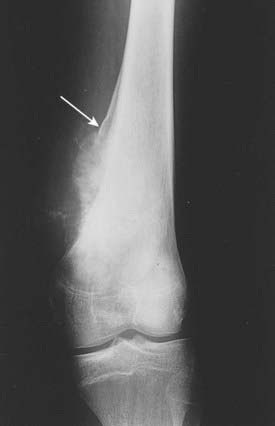
When a malignant condition of bone is suspected, plain film radiographic evaluation will provide, in most instances, a highly reliable presumptive diagnosis and should not be overlooked in the current era of cross-sectional imaging. The age of the patient, location of the tumor within the bone (epiphysis, metaphysis, or diaphysis), and skeletal site (axial or appendicular) set the foundation for radiographic diagnosis. Osteosarcomas commonly arise in the metaphyseal region and may extend into the diaphyseal or epiphyseal region, or both, of the affected bone.1 Cortical bone is destroyed in a disorganized fashion, and the periosteal reaction, which is the effort of the native bone to restore cortical integrity, results in the radiographic appearance of the classic Codman triangle and periosteal bone spicules (Fig. 59-1). The tumor itself images as a cloudlike density representing tumor osteoid. A soft tissue mass is usually present. Magnetic resonance imaging (MRI) is most useful for imaging soft tissue extension as well as identifying intramedullary skip metastases. The latter, which occur rarely, are also well-detected on bone scintigraphy.
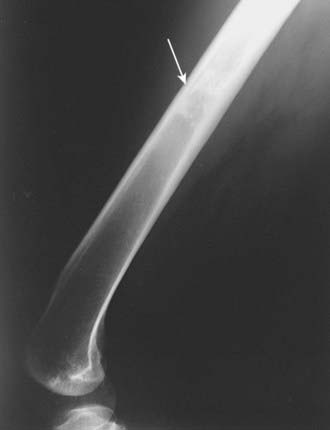
As most cartilaginous lesions are typically slow growing and commonly arise from pre-existing, benign cartilaginous lesions such as an enchondroma or an osteochondroma, the radiographic appearance of a chondrosarcoma may be only subtly different from that of a purely benign condition.2 Although the cartilaginous matrix typically mineralizes in a benign growth, development of lucent areas within the matrix are worrisome for malignant degeneration (Fig. 59-2). Similarly, an enchondroma typically induces endosteal scalloping and gradual cortical remodeling, but when this picture evolves to include cortical destruction and penetration, malignancy is likely. Serial radiographs are recommended in monitoring apparently benign lesions, especially those in an axial location.
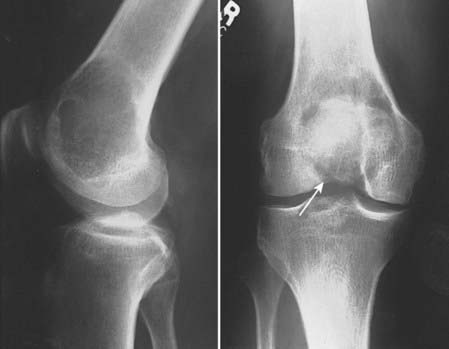
The most important radiographic features of giant cell tumors are their metaphyseal location and rather bland lytic appearance with little or no host response in the narrow transition zone between tumor and native bone.3 Extension to the subarticular cortex in the epiphysis is common (Fig. 59-3). This has given rise to some dispute as to the site of origin, with some investigators citing the epiphysis.4–6 These tumors usually cause cortical thinning, with frank breakthrough being a less common event.
Reports from institutions where the effects of radiation on giant cell tumors have been studied note that bone reparation may not be radiographically evident for up to 2 years after therapy.7,8 The short-term radiographic appearance may actually be confused with tumor progression as the original sclerotic rim involutes. Therefore, patience is required in the interpretation of radiographic studies in the short term after irradiation for giant cell tumors of bone.
Pathologic Features
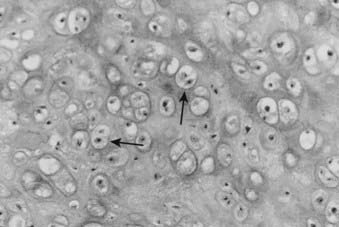
For example, in low-grade chondrosarcomas, mitoses are rare and the appearance of chondrocytes with a plump nucleus or moderate numbers of multinucleated cells may be diagnostic (Fig. 59-4). These histologic findings in association with an axial location in an adult would be more worrisome, whereas they would be of no particular importance in a child or in a distal location. As 10% of chondrosarcomas arise in pre-existing, benign cartilaginous growths, extensive specimen sampling is necessary to avoid overlooking areas of malignant degeneration. The periphery of the lesion is usually most revealing. Although the cartilaginous matrix may contain calcifications and ossifications, malignant osteoid is not seen.
Conversely, malignant osteoid is the hallmark of osteosarcoma. Although it is logical to assume that the tumor arises from osteoblasts, the presence of other malignant mesenchymal tissues within osteosarcomas suggests that the precursor cell is actually of a more primitive mesenchymal, pluripotential origin. Therefore, the microscopic appearance is that of a sarcomatous stroma directly forming tumor osteoid or bone (Fig. 59-5). A large amount of atypical cartilage or fibrosarcomatous matrix may also be evident.
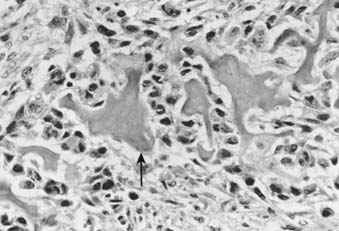
FIGURE 59-5 • Osteosarcoma. These highly atypical cells make up the sarcomatous stroma adjacent to malignant osteoid (arrow).
The physaliferous cell is the histologic hallmark of the chordoma. These so-called bubbly cells have abundant intracytoplasmic droplets of mucoid material and are usually seen in clusters surrounded by a sea of mucoid material (Fig. 59-6). They arise from the remnants of the notochord in the axial skeleton.9 During embryonic development, the notochord undergoes gradual obliteration within the vertebral bodies during the second month of gestation. However, the central portion of the intervertebral disk, which contains notochordal tissue in the form of nucleus pulposus, may persist for an indefinite period.
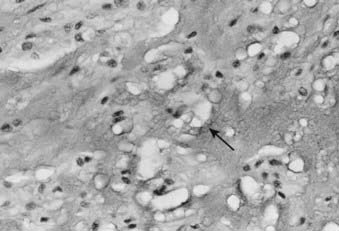
FIGURE 59-6 • Chordoma. The physaliferous cells (arrow) display a bubbly cytoplasm caused by copious intracytoplasmic mucoid droplets.
Experimentally, chordoma-like lesions have been produced in rabbits by piercing the anterior intervertebral ligaments with a needle and allowing the escape of nucleus pulposus.10 In fact, 50% of patients with sacrococcygeal chordomas have a history of previous lower back trauma.11
Radiotherapy Background, Role of Radiotherapy, Technique
Osteosarcoma
Background
True malignant bone tumors represent less than 1% of all malignant neoplasms.12 Osteosarcoma is the most prevalent primary malignancy of bone; it is twice as common as chondrosarcoma, three times as common as Ewing sarcoma, and 10 times more common than malignant fibrous histiocytoma (MFH) of bone.13
Although osteosarcoma can occur at any age, nearly 60% of patients are between 10 and 20 years of age. Not surprisingly, the age peak is younger in females (17 years) than in males (18 years), mirroring the period of most active skeletal growth for each sex. An additional 10% of cases occur in patients older than 60 years of age. There is a slightly greater incidence in males, 1.3 : 1 at Memorial Sloan-Kettering Cancer Center (MSKCC) and 1.6 : 1 at the Mayo Clinic, perhaps reflecting the longer duration of skeletal growth in males and the greater skeletal bulk. Similarly, the distal femur and proximal tibia account for more than 50% of cases in adolescents, as this is the site of most rapid skeletal growth. Until epiphyseal closure, the long bones are involved in nearly 80% of cases, whereas after skeletal maturity, the incidence is approximately equal between the long and flat bones, with the skull and axial skeleton representing 40% of cases.13
In patients older than 60, 56% of osteosarcomas are believed to arise as a consequence of other bone conditions, such as Paget disease and fibrous dysplasia, or following irradiation. In this setting, response to chemotherapy is less likely,14 with a 5-year survival rate of 15% in a review from MSKCC9 and 10% at the Mayo Clinic.15
In a cohort of 900 patients followed at the National Cancer Institute (NCI) Epidemiology Branch who had received repeated injections of radium-224 for the treatment of bone tuberculosis or ankylosing spondylitis, a 200-fold increased risk for developing bone sarcomas was noted.16 The estimated cumulative skeletal dose was 4.2 Gy.
Glicksman and Fabrikant reviewed the current literature regarding postirradiation second neoplasms.17 Bone sarcomas were the most common second malignancy reported. The authors noted that although doses ranged between less than 10 Gy and 80 Gy, only 21% of the secondary sarcomas were along the central axis of the radiation field and nearly 50% were on the field periphery. An additional 30% were located in the superficial tissues, superficial to maximum dose (dmax), causing the authors to speculate that the most likely regions of sarcoma induction received between 20% and 70% of the midplane dose.
Role of Radiotherapy
At least 50 years ago, it was evident that amputation alone was inadequate treatment for the majority of patients with osteosarcoma.18–21 The fact that 80% of patients developed lung metastases within 2 years of diagnosis led to efforts to approach this disease in consideration of its likely systemic nature. In the classic report by Cade in 1955,22 doses of 70 to 90 Gy in 8 to 12 weeks were delivered to the primary tumor at diagnosis and those patients who were free of systemic metastases at 6 to 9 months underwent amputation. Cade’s goal was to spare the children who were destined to develop pulmonary metastases from having an amputation. Among those patients who eventually underwent amputation after the course of radiation, many were found to have complete clearance of histologically viable tumor. Similarly, in 1973, Allen and Stevens23 reported on 10 patients with nonmetastatic osteosarcoma treated with 79 to 100 Gy with either 60-Co or 2-MV photons preoperatively. No histologic evidence of tumor was found in six of seven surgical specimens, and 6 of 10 patients were free of disease at a minimum follow-up of 30 months. The observation of improved survival compared with historic control subjects prompted the authors to speculate that tumors irradiated preoperatively were less likely to cause systemic metastases.
In 1975, Rosen and colleagues at MSKCC24 proposed using high-dose methotrexate with vincristine, doxorubicin (Adriamycin), and cyclophosphamide on the “T-4 protocol” as adjuvant therapy following surgical ablation of primary osteosarcoma. This study was based on their promising results in children with metastatic disease, in which they found a median survival of 3 months without therapy and 15 months with the chemotherapy regimen and an overall response rate of 77%. They argued that local radiation followed by amputation for those patients who had not developed pulmonary metastases was no longer a tenable approach; rather, immediate amputation followed by adjuvant systemic chemotherapy held more hope for the majority, who were assumed to have microscopic dissemination at diagnosis.25
The clear benefit of chemotherapy in an adjuvant setting led Rosen and colleagues at MSKCC to advocate using chemotherapy in a neoadjuvant fashion, thus permitting an “in vivo sensitivity test.”26,27 A good response, it was reasoned, would improve the opportunity to achieve better limb preservation with complete extirpation of the tumor and, moreover, could guide the selection of chemotherapy in the postoperative period.28,29
Recent publications have identified a small group of patients for whom local adjuvant radiation therapy may, in fact, be useful. Ozaki and colleagues,30 reporting for the Cooperative German/Austrian Osteosarcoma Study Group reviewed 1982 consecutive patients entered prospectively onto neoadjuvant chemotherapy studies between 1979 and 1998. Sixty-seven patients were identified with nonmetastatic, high-grade primary tumors of the pelvis (ilium, acetabulum, pubis, and ischium); sacral primaries were excluded. Eleven patients received radiation in some form to achieve local control: four postoperatively, and seven as definitive local management with doses between 56 and 68 Gy employed. The overall survival was statistically superior for this group of 11 patients when compared with 19 patients undergoing similar surgical management (intralesional surgery or no surgery) without radiation. The group of patients irradiated had a 29% 5-year overall survival compared to zero for the unirradiated group (P = 0.0033). The authors conclude that unresectable osteosarcomas or those operated on with inadequate margins should be treated with a regimen including radiotherapy.
Other investigators have noted that primary tumors of the pelvic bones pose a particular technical challenge in achieving a wide local resection while respecting the anatomic requirements to preserve structural and functional integrity.31 Tumors in the sacral and para-acetabular regions especially may benefit from preoperative, intraoperative, or postoperative radiation, either alone or in combination.32,33 Stea and colleagues34 at the NCI contend that the small cell variant of osteosarcoma may be as radiosensitive as Ewing sarcoma. They present both in vitro and clinical evidence for the radiation responsiveness of this tumor subtype and report 100% local control for five patients with gross disease treated with radiation alone.
Osteosarcoma of the axial skeleton presents considerable challenges in efforts toward local control. At the Mayo Clinic,21 patients with osteosarcoma of the spine were treated with radiation after biopsy or decompression and all but one were dead of disease at a median of 10 months.35 Only five patients received chemotherapy in this series. In a review from MSKCC of osteosarcoma of the spine, 13 patients underwent limited resection followed by radiation, whereas 11 patients, treated after 1978, underwent aggressive surgical resection, chemotherapy, and local radiation.36 In the latter group there were five long-term survivors, and aggressive multimodality therapy was recommended.
Investigators from Russia reported that fractionated external-beam radiation to a median dose of 60 Gy (range 40 to 68 Gy) was used after neoadjuvant chemotherapy in 31 patients who refused amputation for extremity osteosarcomas.37 The overall survival, progression-free survival, and metastasis-free survival at 5 years were 61%, 56%, and 61%, respectively. Survival was far higher in those that had a pronounced response to chemotherapy (91% versus 35%). Thus radiotherapy is an option for patients with excellent response to chemotherapy. Two recent reports indicate that postoperative radiotherapy improves results for patients with positive margins.38,39
Osteosarcomas of the facial bones appear to have different biologic features and natural history than those located elsewhere in the body. Numerous reports on small numbers of patients seem to consistently point to a lower tendency for distant metastases. Investigators from the Institut Gustave Roussy40 obtained a clinically complete response after treating a young patient with osteosarcoma involving the nasal cavity, ethmoid, and maxilla with 45 Gy local radiotherapy followed by three cycles of high-dose ifosfamide. Radical ethmoid-maxillectomy revealed 100% tumor necrosis. Suit reported that high-dose radiation followed by resection was used to treat mandibular osteosarcomas with a resultant 5-year disease-free survival of 73%.41 Giuffrida and colleagues42 reported on two patients with osteosarcoma of the mandible treated with preoperative external radiation to 60 Gy, preoperative chemotherapy, resection, and postoperative chemotherapy. Both patients were disease-free at 18 months.
Chondrosarcoma
Background
Chondrosarcoma is a tumor of cartilage cells. It is the second most common primary bone malignancy. This tumor is rare in people younger than 20. After age 20, the risk of getting a chondrosarcoma goes up until approximately age 75. Male/female ratios are 1:1. Chondrosarcomas can develop in any place where there is cartilage. Most develop in bones such as the pelvis, thigh, or humerus. Occasionally, chondrosarcoma develops in the trachea, larynx, and chest wall. Other sites are the scapula, spine, ribs, or skull. A review of the Survival, Epidemiology, and End Results (SEER) database revealed that 2890 cases of chondrosarcoma were reported in the United States between 1973 and 2003. In this group only grade and stage were independent prognostic factors for survival.43
Role of Radiotherapy
The M.D. Anderson Hospital (MDAH) experience with the use of radiotherapy in 20 patients with primary chondrosarcoma of bone for curative intent was reviewed.44 Of 11 patients treated with radiation alone, 9 were controlled for 26 to 156 months. Doses ranged from 40 to 70 Gy in standard fractionation. The investigators believe that radiation is indicated in two settings: (1) as primary treatment when surgery is not possible, and (2) postoperatively when the surgical margins are either grossly or microscopically inadequate. These concepts are in agreement with conclusions drawn from a 23-year experience in the irradiation of chondrosarcoma at the Princess Margaret Hospital (PMH).45,46 No patient had complete surgery, and the majority presented with pain and tumors in an axial site—all poor prognostic features. Despite these facts, 50% of those whose tumors were irradiated with curative intent were controlled locally following treatment, and 25% were free of disease at 15 years. The majority received 50 to 55 Gy in 2.5-Gy fractions. The investigators point out that tumors in the head and neck and truncal regions have an 85% local recurrence rate with surgery alone, supporting the value of radiation in an adjuvant setting where surgical resection is judged to be inadequate.
Huvos and colleagues46 reported a clinicopathologic analysis of 35 patients treated at Memorial Hospital for the mesenchymal variant of chondrosarcoma. They separated this group into patients with a predominant hemangiopericytomatoid component and those with a small cell undifferentiated pattern. These groups demonstrated responsiveness to both chemotherapy and radiation, in contrast to results with conventional chondrosarcoma.
Low-grade chondrosarcomas of the base of the skull have been treated with fractionated proton radiation therapy with a 5-year local control rate of 82%.47 Investigators from the Proton Therapy facility in Orsay, France, reported the results of combined proton-photon irradiation for 45 patients with base-of-the-skull chordomas or chondrosarcomas.48 The median dose to gross tumor was 67 cobalt gray equivalent (CGE) with one-third of the dose being delivered with protons. Three-year local control and survival rates of approximately 90% were reported. Two patients experienced serious late symptoms from treatment—one had memory decline and the other had bilateral vision loss.
The series from Massachusetts General Hospital (MGH) has published the results on treatment of 519 patients with chordomas and low-grade chondrosarcomas treated with proton therapy.49 Five-year local control rates of 73% for chordomas and 98% for chondrosarcomas were achieved with tumor doses from 66 to 83 CGE. Results from Loma Linda show similar success, with 5-year local control rates of 76% for chordomas and 92% for chondrosarcomas.50 Recently preoperative and postoperative radiotherapy has been used employing 20 Gy preoperatively and 50 Gy postoperatively. Local control was reached in 90% of initial cases.51
Technique
Proton beam or IMRT techniques should be employed for chondrosarcoma. Lesions in resectable locations with margins will not require radiotherapy. Thus lesions with close or positive margins located at the skull base, in the spine and sacrum will be the targets. Doses of 50 Gy to subclinical disease, 70 GyE to microscopic disease and 77 GyE to gross disease are advised. Using standard fractionation. Doses to the center of the cord and brainstem must be kept below 54 GyE in 1.8-GyE fractions.52
Chordoma
Background
According to Huvos,9 50% of chordomas arise in the sacrum, 35% in the clivus, and 15% in the true vertebrae. Between 5% and 43% eventually metastasize,9,53,54 with the lungs being the most common metastatic site. Prolonged survival, even with metastatic disease, is not uncommon. Keisch and associates,55 in their report on 21 patients treated at the Mallinckrodt Institute of Radiology, noted that no patient was controlled, regardless of initial therapy. The group treated with conventional radiation alone did most poorly, with no survivors. Patients with lumbosacral tumors treated with surgery plus radiation had a longer mean disease-free survival (6.6 years) than those treated with surgery alone (4.1 years). They recommend doses of 55 to 60 Gy in standard fractionation to respect normal tissue tolerance.
Role of Radiotherapy
Fagundes and associates54 reported a series of 204 patients with chordomas of the base of the skull and cervical spine treated with combined proton and photon radiation to a median dose of 70.1 CGE. Overall, 29% (60 patients) experienced local relapse. Although 10 of 60 who failed locally also recurred distantly, only 2 of 144 patients who were locally controlled developed distant metastases. Local control, therefore, proved to be a highly significant predictor of distant disease-free survival. Overall survival following local relapse was poor, with a 5% actuarial 5-year survival rate and no survivors at 7 years.
Complete surgical excision as the primary procedure with negative margins can be curative, but it is rarely possible. The poor long-term survival after local recurrence highlights the importance of a concerted approach by the surgical and radiation oncologists to achieve local control with combined modality therapy at the outset when negative margins cannot be attained. Local recurrence is the rule when the tumor is violated during resection, and a second radical resection will rarely achieve long-term local control. Heroic surgical procedures, such as hemicorporectomy,56,57 have been advocated for locally extensive growths. However, if positive margins or gross residual disease are known to exist after the primary surgery, postoperative radiation therapy is indicated.
Technique
Adequate delivery of high-dose conventional photon irradiation is virtually impossible owing to the proximity to normal neural tissues and, in the sacrococcygeal region, to the bowel. Therefore, results with conventional photon irradiation have been unimpressive.58 In contrast, proton beam therapy, using precise positioning and immobilization, is able to deliver doses as high as 74 CGE to chordomas and chondrosarcomas of the base of the skull and the cervical spine while respecting the normal tissue tolerance of the brainstem, spinal cord, optic structures, and temporal lobes. Investigators from MGH58,59 stress the importance of extreme positioning accuracy to extract the greatest advantage from the physical dosimetry characteristics of protons.
In addition, promising results with charged particle irradiation (helium and neon) in the treatment of sacral chordomas have been reported by the University of California Lawrence Berkeley Laboratory.60 It is not clear whether heavy charged particle therapy is superior to proton therapy, but there is no doubt that the physical characteristics and radiation biology of these modalities are superior to conventional photon irradiation. An approach using three-dimensional conformal radiation therapy may produce nearly equivalent dosimetric advantages (Fig. 59-7A-D). Such a technique requires fastidious attention to all phases of patient immobilization, fine cut–based computed tomography (CT) or MRI planning, and treatment delivery. For patients with these rare neoplasms, efforts to optimize local control when planning primary management might be best served by early collaboration at a center with such specialized equipment.
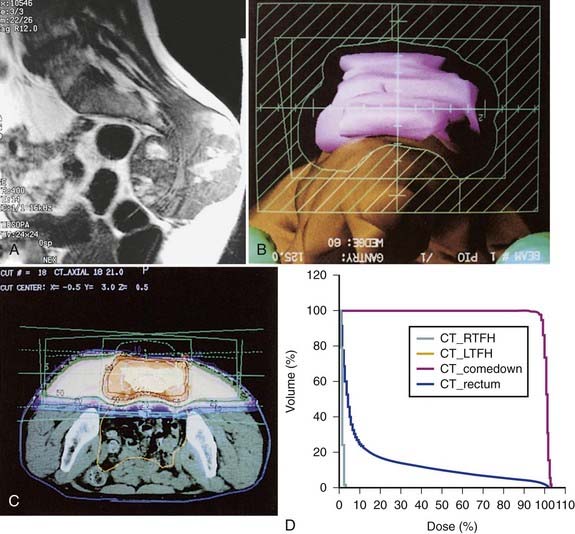
French workers have reported on 100 patients treated with combined photons and protons for tumors of the skull base and cervical spine. Local control rates at 2 and 4 years were 86% and 54%, respectively. Minimum dose level and homgeneity of dose were important outcome-related factors. Median total dose was 67 GyE.61 In a group of extracranial chordomas treated in Switzerland with proton scanning techniques, the median dose was 72 GyE and local control was 86%. Residual tumor greater than 30 cc was a poor prognostic factor.62 Recent results in sacral chordom reported form Boston indicate that local control is high in initial patients with doses of 73 GyE or higher.63 Technique therefore with either protons or IMRT must ensure even tumor coverage, low residual tumor volumes, doses of 72 GyE or higher, and limitation of dose to critical neural structures. Limited results are reported for radiosurgry of skull base chordoma with 5-year projected control in 18 patients of 63%.64
Giant Cell Tumor
Background
Giant cell tumors are more prevalent in females and rarely occur before skeletal maturity is reached, the majority occurring between 20 and 40 years of age. Approximately three-quarters of these tumors occur at the epiphysis of a long bone6 of the extremities, almost always extending to the articular cartilage. Only 3% are in vertebral bodies. Consequently, the majority are managed surgically, with local control rates in the range of 85% to 90%.6
Role of Radiotherapy
Dahlin and colleagues6 reported the treatment results of 195 cases of giant cell tumor of bone treated at the Mayo Clinic between 1910 and 1969. They found the local failure rate to be nearly identical (43%) in primary tumors treated with surgical techniques alone and those treated by surgical removal followed by radiation therapy. The series, collected before the advent of custom allographs and joint prostheses, reveals the necessity of a wide en bloc resection leaving no residual tumor. In the surgery-alone group, 2 of 40 patients developed malignant degeneration at failure, and in the irradiated group, 5 of 61 had this event. Although the authors conclude that radiation is an important factor in the malignant transformation of giant cell tumors, they clearly report the occurrence of (1) malignant giant cell tumors at diagnosis, (2) malignant transformation at recurrence in unirradiated patients, and (3) metastases from “benign” giant cell tumors. Therefore, owing to the inherent selection bias in the aggressive treatment group, the contention that radiation causes malignant transformation is probably untenable. Unfortunately, such reports, when considered along with the young age of most affected patients, contribute to a reluctance to use radiation in many cases.
Series reporting the efficacy of radiation in the management of giant cell tumors always reflect the sites and nature of involvement that are unusual. For example, in the series reported by Chen and associates,8 30 of the 35 cases reported occurred in the skull or spinal axis. In only two cases was radiation delivered after complete surgical excision; 33 patients had gross tumor at the time of irradiation. Patients who received less than 30 Gy experienced local failure in the majority of cases (5 of 8), whereas at doses of more than 35 Gy, the majority (14 of 17) were controlled. Although they present no further dose-response data, the authors do recommend 50 to 60 Gy when radiation will be the sole treatment modality and 30 to 40 Gy in the postoperative setting. In addition, they suggest that a 1- to 2-cm margin be used when designing radiation fields.
Similarly, investigators at the PMH65 report 21 patients treated for 42 years; 8 were treated at recurrence, but of the 13 primary cases, 11 were treated postoperatively for microscopic or grossly positive margins. Of these 11 patients, 10 were controlled with radiation doses between 10.8 and 50 Gy. For the eight patients treated at recurrence, seven were controlled with radiation doses between 35 and 55 Gy. The investigators concluded that even gross disease was readily controlled with moderate doses of radiation. They recommend 35 Gy in 15 fractions in situations in which the probability of recurrence is high and the potential morbidity of further relapse is high. They do not go so far as to recommend that radiation be routinely employed after surgery with resultant positive margins, and, in fact, the results in the recurrent setting were as good as those in the primary setting.
The MDAH group67 reported on 15 patients irradiated for giant cell tumors between 1948 and 1984, of whom 10 were evaluable. Of these 10, 3 patients died of uncontrolled local and distant disease and 5 had received chemotherapy as a component of their treatment course, attesting to the unrepresentative nature of the study population. Radiation doses ranged from 36 to 66 Gy, and the authors recommend 45 to 50 Gy when surgical excision cannot be performed.
In the University of Florida series spanning 15 years,66 all 14 patients were treated for gross unresectable disease; 10 were in the axial skeleton and 6 were recurrent lesions. Nevertheless, 75% were controlled with radiation doses between 35 and 55 Gy, and the remainder were controlled with surgical salvage. The authors recommend doses in excess of 40 Gy.
Aneurysmal Bone Cyst
Background
The aneurysmal bone cyst (ABC) is a solitary lytic lesion of bone arising most often in the second decade and more often in females. The most common locations are the metaphysic of the lower-extremity long bones, vertebral bodies or arches, and the flat bones of the pelvis.68 One theory of their origin is that they are caused by increased venous pressure; another is that they are secondary to trauma or an underlying, usually benign, tumor, often a giant cell tumor. Recently a translocation involving the 17p13 chromosome has been identified in primary ABC but not in secondary types associated with other tumors. These involve CDH11 or USP6 rearrangements and indicate that this is a true neoplasm.70
Role of Radiotherapy
Most lesions can be treated with resection using curettage and a high-speed burr and control achieved in 90% of cases. Rarely, radiotherapy can be used for resistant lesions that threaten to destroy function, such as lesions involving much of the hemipelvis. The University of Florida reported on nine ABC patients treated with megavoltage radiotherapy, most with 26 to 39 Gy. There were no local recurrences.69
Bone Metastases
Technique
Wu et al. performed a meta-analysis of trials published before 2001 and concluded that there was no significant difference in complete and overall pain relief between single and multifraction palliative radiotherapy for bone metastases.71 This conclusion was further strengthened by many other studies. See Chapter 3, “Fractionation Effects in Clinical Practice.”
Epidemiology
An estimated 8300 new cases of soft tissue sarcoma were diagnosed in the United States in 2003 and 3900 died of the disease.72 Soft tissue sarcomas constitute approximately 0.6% of all malignancies diagnosed yearly in this country (excluding skin cancers and carcinomas in situ). They can occur at any age, and, like carcinomas, are more common in older patients. Approximately 15% of affected persons are younger than age 16, and 40% are older than age 55. The male/female ratio is 1.12 : 1.
The pathogenesis of most soft tissue tumors is still unknown. Trauma or past injuries, frequently implicated in the development of sarcomas, appear to be events that call attention to the underlying neoplasm. Rare cases of these tumors arising from scar tissue following surgery, a burn, or the site of a foreign body implantation have been reported.73,74 Sarcomas (usually lymphangiosarcomas) have also been observed in the chronically edematous arm following breast cancer treatment as described in the Stewart-Treves syndrome. Chemical carcinogens, such as dioxin from herbicides and Agent Orange, have been linked to the development of soft tissue tumors. Yet none of the case control studies carried out since 1980 has been able to substantiate the claim.75–77 Olsson et al.78 showed a reduced risk of soft tissue sarcoma development with an odd ratio of 0.57 with the chronic use (>2 years) of oral contraceptives in a population-based study.
Past radiation exposure has been related to the development of soft tissue and bone sarcomas. The interval between radiation and tumor development ranges from 2 to 25 years. The frequency of neoplasm increases with higher radiation dose and longer follow-up.79 Taghian et al.80 reported a 0.2% rate of radiation-induced sarcoma in 7620 women treated for breast cancer. The estimated actuarial frequency of sarcoma development at 15 to 20 years is approximately 0.5%. This number may be slightly higher with the addition of chemotherapy. A U.S. population–based study of 274,245 breast cancer patients showed that those who received breast irradiation had a higher risk of developing soft tissue sarcoma than those not treated with radiation, although the excess risk with radiation was relatively small (incidence of 0.24 with radiation versus 0.14 without radiation at 15 years).81 The most common postradiation soft tissue sarcoma is pleomorphoric unfifferentiated sarcoma, previously called malignant fibrous histiocytoma (MFH)82 followed by fibrosarcoma and malignant nerve sheath tumor.
Genetics, Cytogenetics, and Molecular Biology
Both osseous and soft tissue sarcomas are included in the Li-Fraumeni cancer syndrome, which is characterized by a familial cluster of sarcomas, early breast cancers, brain tumors, leukemia, and adrenal carcinomas.83 Germ line abnormalities of the p53 tumor suppressor gene have been identified in the members of these families.84 Neurofibromatosis type 1 (NF1), characterized by café-au-lait spots and numerous neurofibromas, is another genetic disorder classically linked to the development of soft tissue tumors. Malignant peripheral nerve sheath tumors arise from malignant degeneration of neurofibromas in 1% to 5% of cases.85 The gene for NF1 has been cloned from the pericentromeric region of chromosome 17. The gene product (neurofibromin) appears to have tumor suppressor activities.86 A slight increase in frequency of soft tissue sarcomas has also been found in basal cell nevus syndrome, tuberous sclerosis, Werner syndrome, intestinal polyposis, and Gardner syndrome.
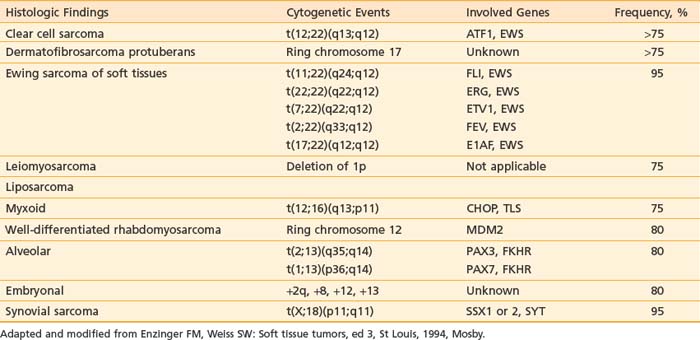
Cytogenetic studies have been carried out for many soft tissue tumors. Table 59-1 lists characteristic chromosomal aberrations and their frequency for certain sarcomas. On the molecular level, alterations in the Rb and p53 tumor suppressor genes have been found in soft tissue sarcomas. Wunder et al.87 reported Rb changes in 5 of 12 high-grade and 1 of 11 low-grade soft tissue tumors. Toguchida et al.88 discovered 42 somatic p53 alterations in 127 cases of osseous and soft tissue sarcomas. Twenty-one were gross gene arrangements, and 21 had subtle changes (missense or nonsense mutations). Others have reported abnormal p53 immunostaining in one-third of the cases.89,90 Of note, mutations of both Rb and p53 genes were frequently seen in the same tumors. MDM2 (located on chromosome 12q13-q14), an inhibitor of p53 transcriptional activities, was found to be amplified in 8 of 24 soft tissue tumors evaluated.91 This suggests that MDM2 amplification is an alternative mechanism for inactivating the cell cycle regulatory pathways. SAS, located in chromosome 12q13-q14 near a cyclin-dependent protein kinase gene, is thought to be involved in signal transduction and cell growth regulation. It was found to be amplified in 7 of 22 MFHs and in three of three liposarcomas.92 Its role as an oncogene has not been clearly established. More details of the cytogenetic changes and molecular biology of soft tissue tumors can be found in an excellent review by Cyril Fisher.93
Transcriptional profiling using either oligonucleotide or complementary deoxyribonucleic acid (cDNA) microarrays has been employed to characterize soft tissue tumors.94 The authors analyzed 41 soft tissue tumors, which provided more than 1.5 million data points for 5520 well-measured genes. Based on the level of gene expression, they found that these 41 specimens can be separated into five distinct groups: synovial sarcomas; gastrointestinal stromal tumors (GISTs); benign peripheral nerve sheath tumors; half of the leiomyosarcomas; and a broad group containing all the MFHs, the liposarcomas, and the other leiomyosarcomas. In addition, they found that c-Kit and protein kinase C are highly expressed in GIST, and cellular retinoic binding protein–1, retinoic acid receptor–γ, and the epidermal growth factor receptor are highly expressed in synovial sarcoma, suggesting that these can be exploited for molecular targeting of these tumors in the future.
Anatomy
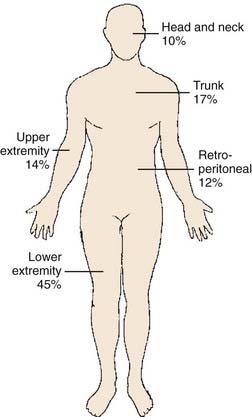
Embryologically, the soft tissue structures arise from the primitive mesenchyme of the mesoderm with some contribution from the neuroectoderm. Soft tissue structures consist of muscles, fat, fibrous tissues, blood vessels, and supporting cells of the peripheral nervous system. The frequency of involvement of different anatomic sites is shown in Table 59-2 and summarized in Fig. 59-8. Soft tissue sarcomas can arise in soft tissues in any part of the body.
Pathologic Conditions
Each of the soft tissues can give rise to a group of malignant sarcomas. Tumor histologic anatomy and their putative cells of origin (as suggested by Enzinger and Weiss85) are listed in Table 59-3. MFH and liposarcoma (Fig. 59-9 and Fig. 59-10) are the most common soft tissue neoplasms in adults, accounting for 35% to 45% of all sarcomas. The three most common sarcomas found in U.S. hospital registries in the 1980s were MFH (26%), liposarcoma (18%), and leiomyosarcoma (15%).95 Hashimoto and colleagues reported an incidence of 25% for MFH, 12% for liposarcoma, and 10% for rhabdomyosarcoma among 1116 cases evaluated.96 More recently, MFH has been renamed and the World Health Organization diagnosis is now called pleomorphic undifferentiated sarcoma (PUS).82
Table 59-3 Histologic Classification of Soft Tissue Sarcoma
| Tissue | Sarcoma |
|---|---|
| I. Fibrous | Fibrosarcoma |
| II. Fibrohistiocytic | Malignant fibrous histiocytoma |
| III. Lipomatous | Liposarcoma |
| IV. Smooth muscle | |
| V. Skeletal muscle | |
| VI. Vascular and lymphatic | |
| VII. Perivascular | |
| VIII. Synovial | |
| IX. Mesothelial | |
| X. Neural | |
| XI. Paraganglionic | Malignant paraganglioma |
| XII. Extraskeletal cartilaginous and osseous | |
| XIII. Pluripotential mesenchymal | Malignant mesenchymoma |
| XIV. Miscellaneous |
MPNST, Malignant peripheral nerve sheath tumor.
Modified from Enzinger FM, Weiss SW: Soft tissue tumors. In Greene LG, Page DL, Fleming ID, et al, eds: AJCC cancer staging manual, ed 6, New York, 2002, Springer, p 193.
MFH was until recently considered the most common soft tissue sarcoma among adults. Histologically, karyotypically, and clinically, this is by far the most heterogeneous group of sarcomas, and the concept of MFH as a histogenetically separate entity was called into question. An alternative hypothesis is that MFH is a poorly differentiated sarcoma and might represent a common endpoint for various other sarcomas. Fletcher et al.82 performed histologic, immunohistochemical, and, where available, ultrastructural reanalysis of 100 MHF cases of the extremity and trunk. In 84 cases, a specific line of differentiation was strongly suggested. The most common diagnosis was myxofibrosarcoma (22 cases) and leiomyosarcoma (20 cases). They also showed that myogenic sarcomas had a worse prognosis than nonmyogenic sarcomas.82 Gene array analysis did not separate MFH from the liposarcomas, which strengthened the alternative view of MFH.94 Further work is needed to clarify this controversy. Even though MFH has been renamed PUS, the term MFH continues to be used and remains useful in diagnostic and clinical practice.
Sarcoma histologic anatomy alone does not provide sufficient evidence for predicting the clinical course. Grading and staging are essential for accurate prognosis and treatment of these neoplasms. Unlike past classifications, each histologic feature can be assigned a specific grade in the current system. When histologic grades are accounted for, with a few exceptions, most soft tissue sarcomas have common biologic behaviors irrespective of histologic type. In a series of 211 high-grade sarcomas, Potter and coworkers showed that histologic anatomy was not a significant determinant for either overall survival or disease-free survival.97
Traditionally, the designation of histopathologic grades depends on a combined assessment of several histologic features: degree of cellularity and pleomorphism, abundance of stroma, expansile or invasive growth, extent of differentiation, mitotic activity, and amount of necrosis. Mitotic count and necrosis appear to be the most significant prognostic factors in predicting the duration of survival and the time to developing distant metastasis.98 There are at least four different grading systems in the literature, as summarized in Table 59-4. Despite their differences, all showed a strong correlation between grade and survival. The two systems most favored by pathologists are those designated as the NCI and the French Federation of Cancer Centers Sarcoma Group (FNCLCC). A direct comparison between these two systems in 410 patients with nonmetastatic soft tissue sarcoma revealed that both systems were of prognostic value in predicting the risk of distant metastasis and tumor-related death, with the FNCLCC system providing a slightly better prediction.99 Unfortunately, there is poor reproducibility of histologic diagnosis and tumor grading among trained pathologists. Coindre et al.100 found a crude agreement rate of only 61% for histologic anatomy and 75% for tumor grade among 15 well-known sarcoma experts.
Clinical Presentation and Routes of Spread
The most common clinical presentation of soft tissue sarcoma is a slow-growing, painless mass. The median period between detection of the lesion and presentation to the clinician is approximately 4 months, and the clinician’s delay in establishing the diagnosis is approximately 1 month.47 Pain, numbness, and swelling may result from tumor invasion of bone or neurovascular bundles.
Approximately 6% to 10% of patients have metastatic disease at diagnosis. Rydholm et al.101 reviewed the records of 278 patients with soft tissue sarcomas of the extremities registered in the Southern District of the Swedish National Registry. They found 19 cases (6.8%) with metastatic disease at diagnosis. In our review of 65 head and neck soft tissue sarcomas treated primarily at the University of California, San Francisco (UCSF), four (6.1%) had distant metastases at presentation.
Hematogenous dissemination after diagnosis is common in high-grade lesions. Isolated pulmonary metastases are the most frequent, accounting for nearly 50% of all initial recurrence.102 Bone, liver, and skin involvement occurred in less than 5% of patients.103 Liposarcoma and retroperitoneal sarcomas display a different metastatic pattern. Although the lungs still predominate as a metastatic destination, liver involvement and peritoneal carcinomatosis do account for a small but significant number of deaths in these patients.102,104 Approximately 75% to 80% of distant metastases appear within 2 years of initial treatment.
Regional lymphatic dissemination is uncommon. In a comprehensive literature review, Weingrad observed a 5.8% incidence of nodal spread during the course of the disease.105 Mazeron found that 5.9% of 323 patients without distant metastatic disease at presentation had nodal involvement.106 There was a correlation between tumor grade and the frequency of nodal metastasis: 0% for grade 1, 2% for grade 2, and 12% for grade 3. Pooled data from published reports on 5257 patients treated for soft tissue tumors showed a higher incidence of nodal spread in certain histologic subtypes: clear cell sarcoma (28%), epithelioid sarcoma (20%), angiosarcoma (23%), rhabdomyosarcoma (15%), and synovial sarcoma (14%).106
Diagnostic and Staging Studies
Radiographic Imaging
Radiologic studies should preferentially be obtained before any surgical manipulation to avoid confusing postsurgical changes. MRI is the preferred imaging modality for soft tissue sarcomas because of its multiplanar imaging capability and superior soft tissue contrast. Most musculoskeletal tumors have intermediate to low signal intensity in T1-weighted images and appear bright on T2-weighted images. Exceptions to these rules are tumors with high fat or blood product content such as liposarcomas and angiosarcomas, which appear as masses of high signal intensities on T1-weighted images (Fig. 59-11).107 The use of contrast agents such as gadolinium has not been shown to improve the accuracy of tumor staging or lesion delineation.
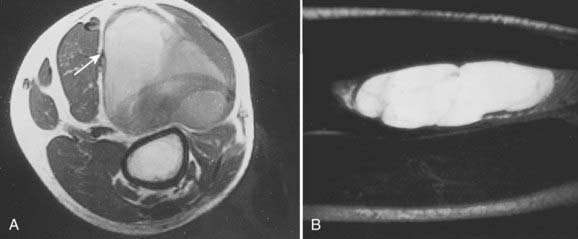
(Courtesy Dr. Charles Peterfly, Department of Radiology, University of California at San Francisco.)
CT remains the diagnostic modality of choice for detecting pulmonary metastases in high-grade tumors.105 Yet the cost-effectiveness of routine chest CT for metastasis screening in patients with T1 and T2 soft tissue sarcoma has been questioned. Fleming et al.108 showed that the routine use of chest CT in addition to a chest x-ray to screen for pulmonary metastasis in patients with T1 tumors of the extremity results in a substantial incremental cost of $2.9 million per case of pulmonary metastasis detected because less than 1% of the patients in this group presented with lung metastasis at diagnosis. Similarly Porter et al.109 found that the use of routine chest CT, when compared with selective CT, for evaluation of metastatic disease for T2 tumors is most cost-effective in patients with extremity and high-grade lesions. For primary tumor evaluation, CT is a useful supplement to plain radiographs to evaluate for suspected bony invasion. Finally, CT is also widely used to image the tumor for three-dimensional treatment planning in radiation therapy.
Bone scans are not useful in the diagnostic workup of soft tissue sarcomas. Bony metastases in the absence of visceral spread are rare. Bony invasion is best demonstrated by CT. Positron emission tomography (PET) scanning and magnetic resonance spectroscopy may help provide information on tumor metabolism and grade,110 although the role of these studies is still undefined. Dimitrakopoulou-Strauss et al.111 studied the use of 18F-FDG PET for the diagnosis of primary or recurrent soft tissue sarcoma and found a sensitivity of 76%, specificity of 43%, and an accuracy of 68%. It appeared to be most accurate in patients with high-grade tumors. Conrad et al.112 reported that the average standardized uptake values (SUV) obtained from PET scans were useful in differentiating high-grade and large tumors with high metabolism from low-grade and small tumors in 108 patients with bone sarcoma and soft tissue sarcoma. Finally, PET may serve as a useful predictor for response from neoadjuvant therapy. Schuetze et al.113 performed serial PET scans on 56 patients treated with two to three cycles of neoadjuvant doxorubicin-based chemotherapy. Patients with a greater than 50% reduction in maximal SUV had a longer time to recurrence (38 versus 18 months, P = 0.03) and overall survival (not yet reached versus 41 months, P = 0.02).