73 Tumor-Targeted Radioisotope Therapy
History of Radioimmunotherapy
In the late 1940s, a method was developed for linking iodine-131 (131I) to proteins, including antibodies (Abs), without significantly altering biologic function, such as Ab immunologic specificity.1 Using this technique, Pressman and Korngold2 demonstrated in 1953 that intravenously administered Ab accumulated to a slightly greater extent in tumors than in normal tissue.3
Clinical trials involving radioimmunotherapy (RIT), or tumor-targeted systemic radiotherapy, began in the 1950s with polyclonal Abs when Beierwaltes4 administered 131I-labeled rabbit Ab to 14 patients with metastatic melanoma and achieved a pathologically documented complete response (CR) in one patient. In the 1960s, Spar and coworkers5 administered 131I-antifibrinogen polyclonal Abs to cancer patients and observed symptomatic improvement in some cases. In 1965, polyclonal Abs began to be developed against specific tumor-associated antigens (e.g., carcinoembryonic antigen). This was associated with greater Ab uptake in tumors. In the 1970s, Ettinger et al.6,7 began to treat patients with cholangiocarcinomas and hepatomas with 131I-anticarcinoembryonic antigen and 131I-antiferritin polyclonal Abs in combination with EBRT (EBRT), doxorubicin, and 5-fluorouracil chemotherapy. A 30% or greater decrease in tumor size was observed in six of nine evaluable patients. Order et al.8 pursued an aggressive combined modality approach to hepatoma, with one trial arm including radiolabeled polyclonal Abs. Because of their polyclonal nature, these tumorphilic radiolabeled Ab mixtures were heterogeneous in their pharmacokinetic and tumor binding properties.
When Kohler and Milstein published, in 1975, a technique9 for the production of monoclonal antibodies (mAbs) in which each clone selected could produce one molecular Ab species of a predefined specificity, this Nobel Prize-winning technology also provided the critical keystone for the development of RIT. With this hybridoma approach, mAbs to predefined cancer-associated antigens could be produced in gram quantities, thus revolutionizing the field. With the exception of anti-idiotypic antibodies, no mAb is absolutely specific for tumor, although tumor to the more sensitive normal tissue radiation dose ratios (therapeutic index [TI]) range from 2 : 1 to greater than 30 : 1 with tumor-targeting radiolabeled mAbs in cancer patients.10,11
MAbs have been conjugated with chemotherapeutic agents, biologic toxins, and radioisotopes in an attempt to use them to selectively target malignant cells while sparing normal tissues.12 Treatment of cancer with mAbs conjugated with chemotherapeutic agents or toxins has faced a number of obstacles, including: (1) the requirement that every malignant cell express the target antigen; (2) the development of multidrug resistance; (3) the degradation of the drug or toxin by lysosomes following endocytosis; (4) the formation of Abs against the toxin; and (5) a dose-limiting capillary leak syndrome manifested by hypoalbuminemia and edema.13,14
Tumor-targeted radioisotopes have several advantages over drugs or toxins: (1) β particles emitted by radionuclides such as iodine-131, yttrium-90 (90Y), or copper-67 (67Cu) can kill adjacent tumor cells (crossfire; Table 73-1), partially overcoming heterogeneous antigen expression and poor tumor penetration; (2) radioisotopes are less subject to multidrug resistance mechanisms; and (3) pharmacokinetic and dosimetric measurements can be obtained from scintigraphic imaging, helping to prevent unacceptable toxicity and optimize the prescribing of therapy.
Radioimmunoconjugates, however, have usually been made with the entire 150 kD mAb, resulting in large molecules that have slow blood clearance, retention in normal organs involved in Ab metabolism, and slow, incomplete tumor penetration. Combined with antigenic heterogeneity, only 0.0001% to 0.1% of the injected dose of Abs binds to each gram of tumor.15 Consequently, RIT has typically produced tumor doses of less than 20 Gy unless bone marrow support allowed larger injected doses. Challenges facing RIT, along with possible solutions,16 are presented in Table 73-2.
Table 73-2 Challenges to Improve the Therapeutic Index for Radioimmunotherapy
| Problems Facing Improvement of the Therapeutic Index | Possible Solution |
|---|---|
| Marrow toxicity | Small targeting molecules |
| Pretargeting approach | |
| Normal tissue antigen | Preinfusion of cold Ab |
| Pretargeting approach | |
| Nonspecific Fc receptor binding | Saturate receptors with cold Ab |
| Intracavitary (e.g., intraperitoneal administration) | |
| Ab fragments: F(ab′)2, svFc, diabody… | |
| Pretargeting | |
| Tumor penetration | Ab fragments: F(ab′)2, svFc, diabody… |
| Multimodality approach: chemotherapy, external beam… | |
| Pretargeting | |
| Inject Ab directly into the tumor |
Ab, Antibody; HAMA, human antimouse antibodies; mAb, monoclonal antibody.
Antibody Structure and Function
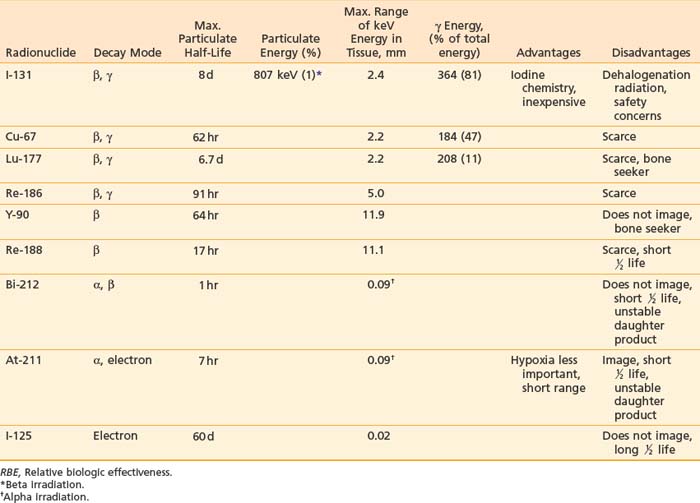
These contain variable and constant regions, with the variable regions of the heavy and light chains (VH and VL) containing the antigen-binding site (Fig. 73-1A). The major constant portion (Fc) of the Ab molecule contains a number of molecular signals that mediate biologic functions such as complement binding, hepatocyte binding, and recruitment or activation of effector cells, such as monocytes and natural killer cells. Pepsin digests of Abs result in a short divalent (F(ab′)2) antigen-binding molecule without most of the Fc fraction, and papain digestion results in an even smaller antigen-binding monovalent fragment (Fab) (see Fig. 73-1A).17,18 Genetic engineering, using the VH and VL MAb DNA, has fashioned smaller binding units, fragment (Fv), as well as the linked single chain fragments (scFvs). These may provide the building blocks for tumor-targeting molecules of the future (Fig. 73-1B).19
Many tumor-binding mAbs can produce antitumor effects: (1) complement-mediated cytotoxicity20; (2) Ab-dependent cellular cytotoxicity21,22; (3) receptor-mediated apoptosis23,24; (4) interference with growth-related receptors25–29; (5) stimulation of the humoral immune system producing a vaccine-like response30–32; or (6) the triggering of apoptosis by appropriate intracellular signal transduction. Smaller binding units (scFv) seldom have these functions, but can be linked into molecular formats developed to improve the TI, such as small high-affinity rapid-targeting radioactive molecules or larger multidentate and multifunctional pretargeted RIT drugs (see Fig. 73-1B).13
Radioisotopes
Many radioisotopes suitable for systemic tumor-targeted radionuclide therapy are presented in Table 73-1. Radioisotopes undergo decay because of the inherent instability of their nuclei. The most important types of decay for medical use lead to γ emission, β and α particles, and electron capture resulting in auger electrons. 131I has both β and γ emissions, the latter being less absorbed by the body, allowing quantitative scintigraphic imaging to determine the pharmacokinetics and calculate the dose to tumors and normal organs from the radiopharmaceutical. By contrast, β particles travel only millimeters to centimeters in tissue and deposit their energy in the vicinity of the point of decay. Beta emissions from radioisotopes such as 131I, 177Lu, 90Y, or 67Cu targeting antigen-positive tumor cells can kill nearby antigen-negative tumor cells through a “crossfire” effect, which increases the homogenicity of tumor radiation.33–36
131I has been used in many clinical trials because of its well-known radiochemistry, availability, and low cost. However, if the radioiodinated MAb is taken into some tumor cells via internalization (endocytosis), the 131I can be enzymatically removed and excreted from the tumor before it can deposit all of its particulate energy, thus, lowering the potential tumor dose. The same process, however, can result in a lower dose to the liver and kidneys since these products, small iodinated peptides and free iodine, are rapidly excreted via the kidneys. It should be noted that free iodine in the blood can be concentrated in the thyroid gland if not blocked in advance by an oral potassium iodine solution. Since the γ rays emitted by 131I have a relatively high energy (364 keV),37 practical considerations are needed to minimize the dose of radiation that is delivered to family members and health care personnel. Patients receiving high doses who are unable to be properly managed at home may need hospitalization to limit the irradiation of others.
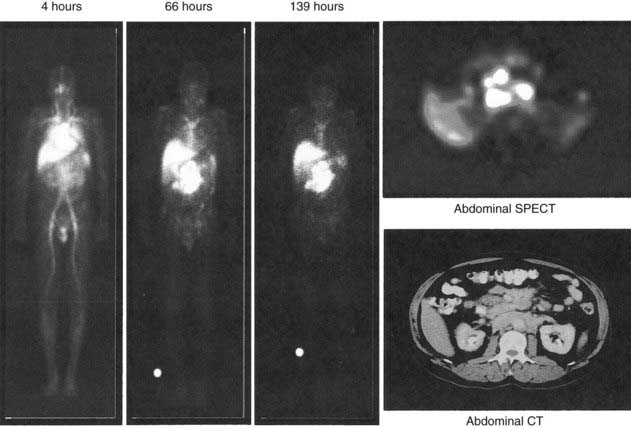
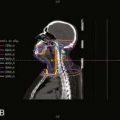
Yttrium-90, a pure β emitter, has three properties that make it an attractive choice for RIT38–40: (1) a high β energy (Eβmax = 2.29 MeV; maximum range of particulate energy in tissue = 12 mm), which enables it to kill adjacent tumor cells and thus, increase the homogenicity of tumor radiation33,34; (2) metal chemistry, which facilitates the presynthesis of storable Ab chelate conjugates that can be quickly and easily radiolabeled in RIT form when needed; and (3) a half-life (2.67 days) that is useful with intact mAbs, but may take 1 to 3 days to concentrate in tumors, compared with smaller fragments and peptides, with maximal concentration within a day. However, 90Y needs well-selected chelation chemistry since free 90Y will accumulate in cortical bone, increasing the radiation to bone marrow. Furthermore, although 90Y-labeled Abs allow easy patient management because they have no primary γ emissions, pharmacokinetics and thus, dosimetry cannot be accurately quantitated by imaging the secondary 90Y emissions. Thus, γ-emitting indium-111 (111In) in the same chelated mAb, when concurrently or sequentially administered, acts as an effective surrogate to provide pharmacokinetic information for its 90Y counterpart (Fig. 73-2).
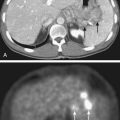
Other radioisotopes have been studied for use in RIT. Generally their availability, cost, labeling, or chemistry characteristics have outweighed the suitability of their physical characteristics for therapy. An example is 67Cu, suitable for RIT because it has 99mTc-like photons for imaging (Fig. 73-3), has a sufficiently long physical half-life for use with intact murine mAb (62 hours), emits β particles similar to 131I, and does not accumulate in bone. Preliminary studies demonstrate 67Cu may provide a better TI than 131I, based in part on its relatively long retention in tumors,41,42 resulting in 1.5- to 5-fold higher tumor doses when equivalent radioactivities (mCi) are administered.43,44 The disadvantage of 67Cu is availability and cost.
Alpha particles deposit energy over a much shorter range than β particles, typically 50 to 100 µm, targeting lesions of 1 to 2 mm diameters (micrometastasis). Thus, radioisotopes that undergo α decay, such as astatine-211 (211At) and bismuth-212 (212Bi) must be within approximately 1 to 2 cell diameters of a tumor cell to kill it.45,46 Although nearby normal cells are spared by α emitters, adjacent nontargeted tumor cells are spared as well. Therefore, these radionuclides are ideal candidates for targeting microscopic residual disease or leukemia, where β particles are relatively inefficient, depositing much of their energy outside their small tumor volume. 211At and 212Bi have been used in clinical RIT trials of leukemia and brain tumors.46,47 The short physical half-lives (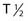 ) of these radionuclides (
) of these radionuclides (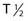 212Bi = 45.6 min;
212Bi = 45.6 min; 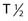 211At = 7 hr) and complex decay cascades has made routine clinical use more difficult (see Table 73-1).45,48
211At = 7 hr) and complex decay cascades has made routine clinical use more difficult (see Table 73-1).45,48
However, the development of new chelators for longer-lived α emitters (e.g., Actinium-225 (225Ac) with 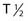 = 10 days) may make future use of RIT with α emitters logistically more feasible and widely available.49 Another promising approach for increasing the efficacy of RIT with α emitters is to link them to small engineered antitumor antibody fragments that are rapidly taken up by targeted tumor. In a preclinical model, diabody molecules were found to be effective agents for targeted radioimmunotherapy of solid tumors using 211At.50
= 10 days) may make future use of RIT with α emitters logistically more feasible and widely available.49 Another promising approach for increasing the efficacy of RIT with α emitters is to link them to small engineered antitumor antibody fragments that are rapidly taken up by targeted tumor. In a preclinical model, diabody molecules were found to be effective agents for targeted radioimmunotherapy of solid tumors using 211At.50
Chelates
Isotopes are attached to mAbs by different methods. The stability of isotope on the mAb is crucial for RIT to maximize the TI. 131I is covalently linked to the Fc portion by iodination of tyrosine residues, whereas radioisotopes of metals such as 90Y, 67Cu, 177Lu, and rhenium-188 (188Re) are more firmly attached by an intermediary molecule known as a chelate. Diethylene-triaminepentaacetic acid (DTPA) was initially used for 111In and 90Y.51 Macrocyclic bifunctional agents48 such as DOTA (1,4,7,11-tetrazacyclododecane-N,N′,N″,N′″-tetraacetic acid) or TETA (1,4,7,11-tetraazacyclotetradecane-N,N′,N″,N′″-tetraacetic acid)52 have increasingly been used because they are useful radiometals with more stable properties in vivo.52–55 For example, stability of yttrium-DOTA minimizes the loss of 90Y to the bone.55
Radiobiology
RIT differs in three important ways from conventional external beam radiotherapy (EBRT) with EBRT using linear accelerators: (1) radiation is continuously delivered to tumors at a low dose rate that initially increases with time as radiolabeled Abs accumulate in a tumor (maximal tumor dose rates approximate ≤0.40 Gy/hr for 90Y-labeled and 67Cu-labeled Abs and <0.10 Gy/hr for 131I-labeled Abs56) and then decreases because of physical decay and biologic clearance35 (compared with a high [∼150 Gy/hr]) constant dose rate intermittently delivered to a limited region of the body with (EBRT); (2) a low dose of radiation is delivered to the whole body; and (3) Abs themselves may, in some cases, exert antitumor effects through biologic mechanisms. For most tumors, one would expect the relative effectiveness of RIT to be approximately 20% less than that of dose-equivalent EBRT, since the low dose rates characteristic of RIT allow time for repair of sublethal damage.56–59 This has not necessarily been the case.
Most in vitro radiobiologic studies of dose rate effects on tumors of 0.40 Gy/hr or less have used nearly constant dose rates.57 In general, these studies have indicated that dose rates of 0.23 Gy/hr or more are required to stop the growth of malignant epithelial cells in vitro,60 although lower dose rates such as 0.09 to 0.11 Gy/hr can halt the growth of HeLa and Morris hepatoma cells61 (dose rates as low as 0.05 Gy/hr can stop the growth of lymphoma cells in vitro).62
Although radiolabeled Abs emit radiation at a low dose rate, several studies of human tumor xenografts in mice have suggested that RIT can, in certain instances, exert greater antitumor activity than dose-equivalent EBRT.63–65 A number of explanations have been proposed to account for an inverse relationship between dose rate and cell killing: (1) reoxygenation during the protracted course of irradiation delivered by RIT, increasing the radiosensitivity of tumor cells66; (2) targeting by RIT of a rapidly proliferating subpopulation of tumor cells (growth fraction) that is largely responsible for tumor doubling67; and (3) low dose rate irradiation that, in some cases, may cause tumor cells to accumulate in the radiosensitive G2/M phase of the cell cycle (G2 block) to a greater extent than conventional dose rate irradiation.68 Furthermore, the importance of sublethal damage repair mechanisms may be minimal for tumors (e.g., lymphomas that radiobiologically have a small shoulder and large α/β ratio),58 and some MAbs have biologic effects that may acutely increase tumor blood flow, elicit an inflammatory or cell mediated immune response, or sensitize tumor cells to the effects of radiation.69 In addition, low-dose-rate irradiation may induce apoptosis to a much greater extent than high-dose-rate irradiation in some tumor types (e.g., lymphoma),70,71 and this effect may be enhanced in a synergistic manner by various biologic effects of some tumor targeting mAbs.
There is evidence that apoptosis is the major response of many tumors to RIT. In experimental human lymphoma tumors (Raji) treated with 67Cu-1-2IT-BAT-Lym-1, apoptosis preceded tumor regression by 4 to 6 days. In these therapy-resistant human lymphoma tumors, apoptosis was convincingly demonstrated to be a major mechanism for the effectiveness of RIT and occurred by p53-independent mechanisms.72 Human breast carcinoma xenografts (HBT3477) have also demonstrated increased apoptotic activity independent of p53 after RIT.73 In addition to the induction of apoptosis, RIT and the associated low dose rate and relatively low dose irradiation can have a variety of important biological effects on tumors, normal tissues and the immune response.74 Potential clinical applications of the low dose rate irradiation from RIT include augmentation of antitumor responses, with possible use in conjunction with tumor vaccines, and radioadaption of normal tissues. Additional studies are needed to optimize the therapeutic utility of both bystander and low dose rate effects in order to maximize the therapeutic index of RIT.74
Linear energy transfer (LET) represents the average energy (keV) locally imparted to a medium by radiation in traversing one micrometer along its path or track.66 Because energy exchanges with matter are widely spaced with low LET radiation, a β particle has only a small probability of releasing enough energy along its track to produce DNA breaks. Approximately 200 DNA double-strand breaks per cell are required to sterilize 99% of a tumor-cell population when low LET radiation is administered.34 The cytotoxicity of low LET radiation may also be diminished by tumor hypoxia.75 In addition, low LET radiation may not be completely tumoricidal because repair of sublethal damage can occur at low dose rates.74
By using a radionuclide that emits β particles with a high energy (e.g., 90Y rather than 131I), dose rates can be increased up to tenfold because of the higher energy deposited by each disintegration of 90Y relative to 131I (see Table 73-1).76,77 High LET irradiation (α emitters) is densely ionizing along particle tracks and consequently is more efficient at producing DNA breaks than low LET radiation. The cell killing produced by high LET radiation is less dependent upon hypoxia77 since it directly produces irreparable DNA breaks, whereas low LET radiation forms highly reactive molecules that produce repairable DNA breaks and relies upon oxygen to prevent repair of these breaks.66 In conclusion, a better understanding of mechanisms and timing of RIT-induced tumor cell death may lead to the selection of combined therapies that enhance tumor responses without increased toxicities.
Dosimetry and Therapeutic Index
The concept of dosimetry is important for treatment planning and the assessment of results. Compared with dosimetry for EBRT, dosimetry for RIT is less precise and is dependent upon (1) the kinetics of uptake and clearance of radiolabeled Abs; (2) the distribution of radiolabeled Abs; and (3) the radioisotope attached to the Abs.78–80
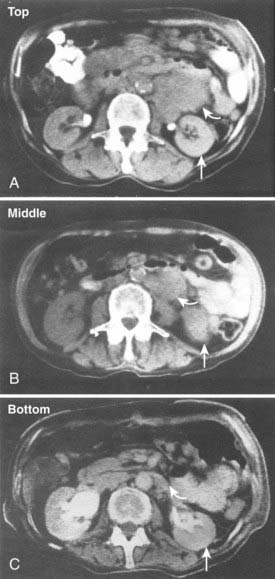
Serial quantitative studies with γ camera imaging (see Fig. 73-2) have traditionally been used to measure radioactivity, determine pharmacokinetics, and calculate dosimetry. Magnetic resonance (MR) and computed tomography (CT) imaging provide normal tissue and tumor volumes.81,82 However, accurate measurement of the radioactivity in small, deep-seated tumors, surrounded by background radioactivity, is difficult. Several excellent overviews of quantitative imaging techniques and approaches to treatment planning in RIT have been published.80,82–85 The medical internal radiation dose (MIRD) method has traditionally been used to calculate absorbed doses of radiation to normal organs, tumor, and the whole body following the administration of radiopharmaceuticals.86 The MIRD method makes a number of assumptions, including: (1) radioactivity is uniformly distributed in that entity, and (2) nonpenetrating, (e.g., β) emissions from radionuclides are completely absorbed in a tumor or organ. These assumptions are reasonable provided that the range of β emissions does not exceed the diameter of the target, which is usually not the case. Specific absorbed fractions (S factors) for penetrating emissions have been calculated for many organs using mathematical anthropomorphic phantoms for a “standard” man, woman, child, and infant that approximate human anatomy. Actual patient organ volumes provide the best alternative.87 For radionuclides that undergo α decay (or electron capture), energy deposition needs to be considered at the cellular and subcellular level,88 which entails the use of a Monte Carlo model89 or analytical microdosimetry with Fourier transform techniques.90
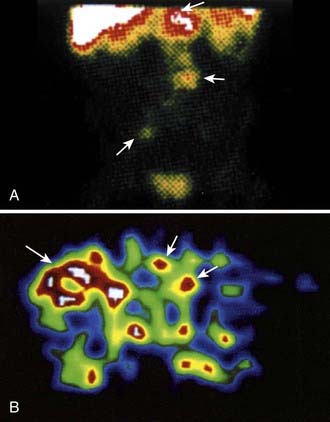
The dosimetric process currently employed in many therapeutic regimens consists of the pretherapy administration of a tracer dose of RIT (or its γ-emitting surrogate) followed by serial planar conjugate view imaging using a γ camera to determine the quantitative spatial distribution of the radionuclide (see Fig. 73-3A). Three-dimensional images from γ camera tomographic scintigraphy (SPECT) provide more detailed pictorial information about deep-seated tumors, but have inherent problems with providing quantitative data (see Fig. 73-3B). Using appropriate standards and planar imaging data, cumulated activity (area under the curve [AUC]), (i.e., the amount of radioactivity integrated over time or residence time), can be calculated for organs or tumor and related to the organ or tumor volume. MIRD “S” values provide the tissue-absorbed fraction for final calculation of absorbed dose to specific organs and tumors.91
Various dosing methods for prescribing treatment doses for RIT have been used (Table 73-3).91 Most clinical trials present their results by reporting the injected activity in millicuries (mCi) or Becquerels (Bq) per body surface area (m2) or weight (kg). One mBq is equal to 2.7 × 10−2 mCi (0.027 mCi). The tumor-absorbed doses are reported either in absolute numbers (cGy or rads) or relative to the injected activity (e.g., cGy/mBq, rads/mCi). Except for intratumoral administration, the average tumor doses delivered over several days in most RIT using nonmyeloablative therapy are 7 to 30 Gy, and in myeloablative regimens, are 30 to 60 Gy.
Table 73-3 Dosing Methods in Radioimmunotherapy Trials
| Dosing Methods | Example |
|---|---|
| Radiation dose (cGy)-based methods/prior dosimetry study | |
| Marrow radiation dose (nonmyeloablative) | 131I-tositumomab |
| Total body surrogate | |
| Blood/body surrogate | |
| Marrow imaging | |
| Critical (dose-limiting) organ radiation (myeloablative) | 131I-tositumomab |
| Radionuclide dose (radioactivity, gBq or mCi)-based methods | |
| Fixed total gBq (mCi) | 131I-antitenascin, 90Y-antitenascin |
| gBq (mCi) per unit body weight (kg) | 90Y-tiuxetan ibritumomab, 90Y-CC49 |
| gBq (mCi) per unit body surface area (m2) | 131I-Lym-1, 131I-hMN14 |
Adapted from DeNardo GL, Juweid ME, White CA, et al: Role of radiation dosimetry in radioimmunotherapy planning and treatment dosing, Crit Rev Oncol Hematol 39:203, 2001.
Approaches to Improve Therapeutic Index
Enhancing the TI is critical to the future success of systemic tumor-targeted radiation therapy, because dose intensification is needed to enhance the efficacy of RIT.92 Circulating radiolabeled mAb irradiates the most sensitive tissue, bone marrow. Marrow toxicity is thus the most frequent dose-limiting toxicity from non–marrow-supported RIT. In an effort to reduce marrow radiation from exposure to hours of circulating radioactive drug, small molecule radionuclide carriers, cleavable linkages of radioactivity, pretargeting, or multistep strategies have been developed that demonstrate exciting potential. These approaches have been applied to increase the TI in both NHL and solid tumors.11–13,93,94
Cleavable Linkers
Organs such as the liver or kidneys are involved in the metabolic pathway of proteins and thus concentrate radiolabeled mAb (in the liver) or small fragments (in kidney tubules). Several strategies have been described to accelerate clearance of radioactivity from these organs and thus to enhance TI.95 These include cleavable linkers, the pretargeting approach previously mentioned to lower blood and marrow dose, and Ab Fvs.
As an example, 90Y DOTA linked to m170 mAb by a peptide preferentially catabolized in the liver resulted in a one third reduction in liver concentration and thus radiation doses in prostate and breast cancer patients, compared to the more conventional DOTA-mAb linkage.96 Importantly, tumor targeting was not diminished (see Fig. 73-3). The results of this study were consistent with in vitro data, which indicated that the peptide linker of DOTA-peptide-mAb is catabolized by cathepsin B, releasing the small radioactive molecule that should be rapidly excreted.95 A variety of concepts have been used to develop linkers to lower this retention of radioactivity in the renal tubule, since small radioactive mAb fragments and peptides have little hepatic uptake and more rapid blood clearance, but usually demonstrate high localization in the kidneys. Zimmermann et al.97 evaluated the peptide-linked copper chelators CPTA-triglycyl-L-p-isothiocyanato-phenylalanine (CPTA-R1-NCS) and the DOTA-triglycyl-L-p-isocyanato-phenylalanine (DOTA-R1-NCS) coupled to F(ab′)2 Fvs. DOTA-R1-F(ab′)2 achieved the best tumor/tissue ratios. Arano et al.98–100 took advantage of lysine-specific carboxypeptidase activity in the tubular cell brush border to develop linkages that appear effective in reducing renal concentration of radiometal from radiolabeled Ab fragments and peptides.
Pretargeted RIT for Dose Intensification
This drug delivery system separates the specific localization of the initially injected larger tumor-targeting mAb from the subsequently injected smaller radioactive molecule, which is quickly bound at the tumor site or rapidly excreted in the kidney. The procedure was first described by Goodwin et al.101 and provides an alternative to increase the therapeutic ratio by selectively targeting the tumor tissue while minimizing the radiation-absorbed dose to normal tissues, particularly blood and marrow, by the excretion of unbound radioactivity. A number of pretargeting systems have been described that use different first-step conjugates94 that can be biotinylated,102 streptavidin conjugated,103 or use bifunctional Abs,104 or oligonucleotide conjugates.105 An important proof of principle pretargeting study for delivering systemic radiation therapy to metastatic cancers has been reported by Breitz et al.106 using a streptavidin/biotin strategy in a three-step pretargeting approach with an anti-CD20 mAb in patients with NHL. Tumor-to-whole body-ratios were obtained that were substantially higher than those achieved with conventional anti-CD20 RIT. In these pretargeting studies using scFv with a streptavidin conjugate, the clearing agent biotin-galactose–human serum albumin was used before injection of radiolabeled DOTA-biotin,107,108 with resultant reduction in circulating Ab by greater than 95%. Weiden et al.11 reported a three-step procedure in patients with NHL. The anti-CD20, C2B8 Ab conjugated to streptavidin (C2B8/SA), was followed by 90Y-DOTA-biotin after a clearing agent; 90Y-DOTA-biotin was tolerated at administrated doses of 29 ± 23 cGy/mCi and mean tumor to whole body ratios of 38 : 1. In six of seven patients with injected activities of 30 to 50 mg/m2 90Y, three CRs and one partial response (PR) were observed. However, high cure rates and high tumor-to-normal ratios in mice,93,109 as well as promising clinical results110 using bispecific Abs (F[ab′]2 fragment) followed by radiolabeled bivalent hapten, have also been achieved without the use of clearing agents.
RIT results in patients reported by Barbet et al.93 using bispecific Abs as the pretargeting agent and subsequent radioiodinated bivalent haptens to bind and crosslink antigens on targeted tumor cells, demonstrated useful effects of modestly increasing radioactive hapten circulation time, with greatly increased tumor retention by crosslinking of the bivalent second agent on the tumor. This bivalent radiolabeled hapten approach increased the tumor-to-blood ratio eight times over previous methods.111
Fractionated Radioimmunotherapy for Dose Intensification
Dose distribution after RIT is known to be highly inhomogeneous, due to the inability of mAb to penetrate uniformly throughout the tumor.112,113 Vascular density, inhomogeneous tumor blood flow, or interstitial pressure are among the related factors that cause an uneven tumor distribution.112,114,115 Single administration of low-dose-rate irradiation may lead to cold spots in the tumor and subsequently to tumor recurrence.116 Several strategies have been proposed to overcome this biologic problem related to mAb.117 The goal of using multiple fractions in RIT is to deliver a more homogeneous distribution of antibody, and therefore radioactivity, with higher associated cumulative radiation doses in tumor and fewer and/or less severe side effects.117,118
There is experimental and clinical evidence that fractionated RIT is effective in different tumor systems. Schlom et al. demonstrated the effect of fractionation in a human colon xenograft using 131I-B72.3 against the TAG-72 antigen. Lethal doses in mice were fractionated to assess the toxicity and the tumor growth effect. One 600-mCi dose produced a mortality rate of 60% in the mice, whereas two 300 mCi weekly doses produced 90% response rates and 10% toxic deaths.119 Dose fractionation permitted further dose escalation in this model. Three weekly doses were allowed with improved results. The same effect was also observed by Buchsbaum et al.120
Clinical experiences with multiple doses of RIT are described earlier in this chapter.121,122 Several important issues must be addressed for optimization of fractionated RIT: (1) decreased immunogenicity of mAb; (2) number of radionuclide doses; (3) dose amount per cycle; (4) interval between doses; and (5) optimal radionuclide physical half-life for a specific treatment interval.117
Clinical Studies
Lymphoma
Since 1985, promising results for RIT in NHL have been described. A summary of the main mAbs (anti-CD20, -CD22, -Lym-1) used in different clinical trials, isotopes, and commercial names are described in Table 73-4. RIT for NHL uses dose regimens that consist of either a single dose of a radionuclide chemically linked to an antibody, at either nonmyelosuppressive or myeloablative dose levels, as well as the use of multiple (fractionated) doses, consisting of dose spacing by days or weeks.
Lym-1
After developing pharmacokinetic data in patients for 131I-mAb on fragments from 1982 to 1984, DeNardo et al.123 conducted an early clinical study of 131I-Lym-1 in patients with advanced lymphoma (Fig. 73-4). Substantial responses in most of the first 10 patients124 led to expanded studies and studies by others. DeNardo et al.125 have summarized their results in patients with B-cell malignancies from the past two decades. Two consecutive trials have been reported using fractionated RIT. Thirty patients with relapsed B-cell malignancies were treated with repeated cycles (10 to 100 mCi) of 131I-Lym-1 as part of a “low-dose fractionated” phase I/II study. PR was achieved in 17 patients (57%; 13 NHL patients and four CLL patients). Advanced disease often interrupted therapy prematurely. However, 18 NHL patients who received at least 180 mCi of 131I-Lym-1, 94% responded to the therapy.121 In contrast there were no responses in patients previously treated with unconjugated Lym-1,126 which also had no dose-limiting toxicity. In a second phase II trial designed to find the maximum tolerated dose (MTD) for at least the first two of up to five doses of fractionated RIT, which binds the β subunit on HLA-DR10; 20 patients with relapsed or refractory NHL were treated in cohorts in a dose-escalating study with 131I-Lym-1 (40 to 100 mCi/m2 at 4-week intervals). The response rate was 71% for those who received at least two doses of 131I-Lym-1. Seven of the responses were complete, with a mean duration of 14 months. All three patients in the 100-mCi/m2 cohort had complete remissions. Responses were observed in all histologic grades and were more rapid and complete when higher doses of radioactivities of 131I-Lym-1 were delivered; however, myelosuppression occurred earlier and was more severe.122 In general, two or more 80-mCi/m2 intravenous infusions of 131I-Lym-1 at 4-week-intervals were well tolerated.
A phase I study for NHL patients with 67Cu-2IT-BAT-Lym-1 resulted in a 58% (seven of 12) overall response rate (ORR).127 For a given level of administered radioactivity, higher absorbed doses of radiation were achieved in the tumors with 67Cu-2IT-BAT-Lym-1 than 131I-Lym-1. When Lym-1 was labeled with 90Y (90Y-2IT-BAD-Lym-1), five of eight patients that failed previous chemotherapy had a PR or stabilization of NHL after RIT.128
Anti-B1 (131I-Tositumomab, Bexxar)
Several phase I/II studies assessed the efficacy and safety of 131I-tositumomab. Kaminski et al. treated 59 patients with refractory/relapsed NHL in a phase I/II single-center study.129 Fifty-three patients received individualized therapeutic doses, delivering a specified total-body radiation dose based on the patient-specific clearance rate of a preceding dosimetric dose. Unlabeled Ab was given before labeled dosimetric and therapeutic doses to improve biodistribution. Forty-two (71%) of 59 patients responded and 20 patients (34%) had a CR. Thirty-five (83%) of 42 patients with low-grade or transformed NHL responded, versus seven (41%) of 17 patients with de novo intermediate-grade NHL (P = .005). The median progression-free survival for the 42 responders was 12 months and 20.3 for those with CRs. Reversible hematologic toxicity was dose limiting. Only 10 patients (17%) had human anti-mouse Abs detected. Similar information was provided in a multicenter trial reporting a 57% response rate and 32% CR rate.130 Both studies demonstrate that a single dose of iodine 131I-tositumomab can produce frequent and durable responses in low-grade or transformed NHL, with a very acceptable toxicity profile.
A pivotal multicenter clinical trial with 131I-anti-CD20 (131I-tositumomab) in patients with low grade or transformed low grade NHL (who had failed previous chemotherapy) compared responses and duration of response in 60 patients to that of their response to their last chemotherapy regimen (LCR).131 After 131I-tositumomab, a response was observed in 39 patients (65%; with 81% response rate in patients with low grade histologies, and 39% response rate in patients with transformed low grade NHL), compared with responses in 17 patients (28%) after their last chemotherapy regimen (P < .001). The median duration of response and CR rates were 6.5 months for 131I-tositumomab compared to 3.4 months for the LCR (P < .001), and 17% CR after 131I-tositumomab (P < .001), compared to 3% after LCR. The median duration of response for CR was 6.1 months after the LCR, and had not been reached with follow-up of more than 47 months following 131I-tositumomab treatment. Another study compared the relative contribution of the antibody (tositumomab) and radionuclide (131I) to both efficacy and toxicity in a randomized two-arm open label study, with unilateral cross over of patients treated with unlabeled MAb to the RIT arm at the time of disease progression.132 On Arm A (RIT), patients received the 131I-tositumomab 450 mg unlabeled MAb with 131I (5 mCi) on 35 mg MAb on day 0. On day 0, patients on Arm B received 485 mg tositumomab (the same mg amount of total MAb as Arm A). Following imaging studies in Arm A, patients were treated on day 7 with 450 mg unlabeled MAb and 131I (dose estimated to deliver 75 Gy to the whole body) on 35 mg of MAb. On day 7, patients in Arm B received 485 mg of tositumomab. The overall response rate was 55% for Arm A and 17% for Arm B (P = .001), with CR rates of 33% and 8%, respectively (P = .012). The median duration of response was 7.6 m for Arm B and had not been reached in Arm A at the time of publication. Hematologic toxicity was more profound in Arm A, with 33% of patients experiencing grade III or IV neutropenia and thrombocytopenia, compared with 8% and 0%, respectively for Arm B.132
Other studies have also demonstrated benefit from retreatment with 131I-tositumomab, 133 as well as the use of 131I-tositumomab in rituximab failures,134 with an overall response rate of 65%, CR rate of 38%, and median progression-free survival of 24.5 months for responders. Kaminski et al.135 performed a Phase II trial of 131I-tositumomab in 76 untreated patients with newly-diagnosed, low-grade B-cell NHL. The overall response rate was 95%, with median duration of response of 71.6 months. The CR rate was 75% with the median duration of CR not reached at the time of publication. Five-year progression-free survival was 59% with a median follow-up of 61.6 months. In contrast to previously-treated patients, no supportive care was needed, and 65% of patients made a human antimouse antibody (HAMA) response, which would most likely preclude future treatment with murine or chimeric antibody, and thereby raises an important question about the optimal time for RIT with immunogenic antibodies in the natural history of NHL.
Given the efficacy and favorable toxicity profile (reversible myelosuppression and generally mild nonhematologic toxicity) in the studies reported above, recent studies have been initiated to investigate the utility of 131I-tositumomab: (1) in combination with chemotherapy in the adjuvant setting, (2) in other histologic types of NHL (e.g., diffuse large cell lymphoma [DLCL]), and (3) combination with EBRT. A phase II trial of cycloposphamide doxorubicin vineristine prednisolone (CHOP) combined with 131I-tositumomab (SWOG 9911) in newly-diagnosed patients with follicular NHL resulted in a response rate of 80% with 52% CRs.136 These encouraging results provided a compelling rationale for an ongoing phase III trial (SWOG 0016) initially comparing CHOP, CHOP + rituximab, and CHOP + 131

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


