Fig. 18.1
Molecular structure of antibodies. Antibodies of the IgA, IgD, IgE, and IgG have two antigen-binding sites. IgA antibodies can be dimeric and IgM are pentameric. Monomeric antibodies have two antigen-binding sites per molecule. Antigen-binding sites are located in the complementarity determining regions (CDRs) within the Fab portion of the antibody. The Fab part, which stands for “antigen binding,” is composed of domains associated with the light chain (VL, CL) and domains associated with the heavy chain (VH, CH1). The Fc portion of the antibody contains the CH2 and CH3 domains of the heavy chains. This region of the antibody can bind to a wide range of cell-associated receptors (e.g., Fc receptors)
The first therapeutic antibodies were obtained by immunizing large animal (e.g., horses) against the targeted antigen (e.g., diphtheria toxin), resulting in the production in the serum of large, polyclonal antibodies specifically recognizing multiple epitopes of the antigen. Then the serum of these animals was used to treat patients (e.g., diphtheria). These polyclonal antibodies were responsible for many adverse events in patients, mainly because of their allogeneic nature that could be recognized and rejected by the host immune system.
Köhler and Milstein described in 1975 the hybridoma technique which allows to generate antigen-specific monoclonal antibodies [5]. This technique consisted in vaccinating rodents (mice, hamsters, etc.) with a specific antigen and subsequently collects the B cells from the spleen of the immunized animals. These B cells were then fused with an antibody nonproducing plasma cell cancer cell line (myeloma). These fusions between a primary B-lymphocyte and a myeloma cell are called “hybridoma.” The mixture of hybridoma cells, made of multiple different primary B-cell clones, each secreting one specific antibody, could be separated into individual single cell culture wells. Those wells can then be screened to identify the ones which contain the hybridoma secreting the desired antibody. Hybridomas can be expanded in vitro in culture, and antibodies can be concentrated and purified from the culture supernatant.
Since the hybridoma technique has been described, numerous other techniques have been developed, generally with the aim of not only improving the yield of monoclonal antibodies production but also reducing the immunogenicity of monoclonal antibodies [6]. Indeed, rodent-derived monoclonal antibodies can be recognized as foreign by the human immune system which can generate human anti-mouse antibodies (HAMA). Patients developing high levels of HAMAs can increase the clearance rate of the monoclonal antibody from their blood-reducing exposure/efficacy. HAMAs can also be responsible for serious anaphylactic or anaphylactoid adverse reactions. Therefore, many strategies have been developed to reduce the immunogenicity of rodent monoclonal antibodies, which have allowed to generate chimeric murine/human monoclonal antibodies or humanized monoclonal antibodies and eventually fully human monoclonal antibodies. The degree of humanization of monoclonal antibodies is specified in their name (Fig. 18.2). Chimeric monoclonal antibodies contain the rodent heavy and light variable chains from the Fab component, thereby retaining the antigen-binding specificity of the initial rodent monoclonal antibody. These variable parts are genetically fused to the human immunoglobulin constant regions (constant Fab and constant Fc regions). This chimerization was a way of preserving the specificity and affinity of the antibody while reducing its immunogenicity in immunocompetent patients. Humanized monoclonal antibodies consist in grafting human framework regions into the variable regions in order to only retain the murine complementarity-determining regions (CDRs) which have the antigen-binding ability. Fully human monoclonal antibodies have been generated, thanks to transgenic mice bearing human immunoglobulin genes. These mice can therefore produce fully human monoclonal antibodies upon immunization. Fully human monoclonal antibodies have significantly reduced immunogenicity and therefore generate low levels of human antihuman antibodies (HAHAs). The low titers of HAHAs usually do not impact significantly the pharmacokinetics of the human monoclonal antibodies and generate low levels of infusion-related reactions (<5% of patients) [7].
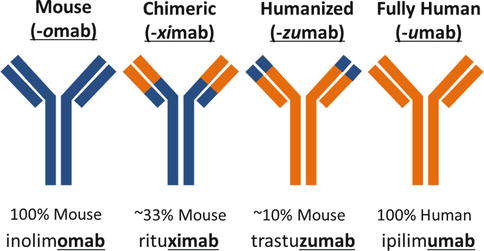
Fig. 18.2
Nomenclature and significance of monoclonal antibody names. As opposed to murine or chimeric monoclonal antibodies, humanized and fully human antibodies are poorly immunogenic and are therefore better tolerated and have better pharmacokinetic profiles
18.3 Monoclonal Antibodies, a Versatile Platform for Cancer Therapies
In 1982, Levy et al. demonstrated for the first time that tumor responses could be obtained in patients with B-cell lymphoma after administration of an antitumor monoclonal antibody designed to target specifically the BCR of their B-cell malignancy [2]. These first therapeutic monoclonal antibodies have inaugurated the concepts of tumor-targeted therapy and personalized medicine. The limitation of this approach was that it needed the generation of a personalized anti-BCR monoclonal antibody for each patient. In order to circumvent this limitation, a monoclonal antibody was generated against CD20, an antigen commonly expressed by B cells and therefore by B-cell lymphomas. This monoclonal antibody, called rituximab, became the first monoclonal antibody to gain FDA approval for the treatment of cancer. Since then, many other tumor-targeting monoclonal antibodies have been approved in oncology and other indications (Fig. 18.3). The most prescribed tumor-targeted antibodies are directed against CD20 in B-cell malignancies (e.g., rituximab), against HER-2 (e.g., trastuzumab) in HER2-positive breast cancers, and EGFR (e.g., cetuximab) in head and neck squamous carcinomas and RAS wild-type colorectal cancers. All types of antigens could be in theory targeted by antibodies, including non-peptide ones such as gangliosides. Gangliosides are molecules composed of a glycosphingolipid (ceramide and oligosaccharide) with one or more sialic acids linked on the sugar chain. Recently, it has been demonstrated that a monoclonal antibody targeting the ganglioside GD2 could improve the survival of children with high-risk neuroblastoma [8].
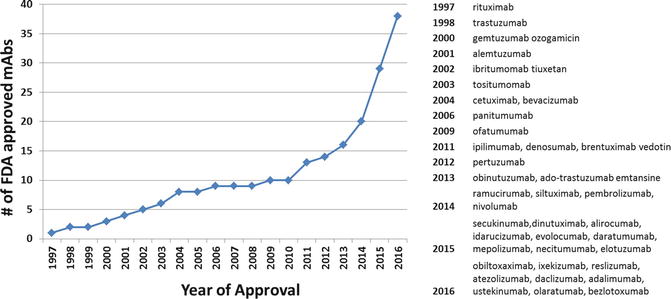
Fig. 18.3
FDA approvals for monoclonal antibodies. Monoclonal antibodies developed for autoimmune or inflammatory disorders could be of interest for treating immune-related adverse events of immune-targeted antibodies. Molecular targets: rituximab, CD20; trastuzumab, HER2; gemtuzumab ozogamicin, CD33; alemtuzumab, CD52; ibritumomab tiuxetan, CD20; tositumomab, CD20; cetuximab, EGFR; bevacizumab, VEGFR; panitumumab, EGFR; ofatumumab, CD20; denosumab, RANKL; brentuximab vedotin, CD30; ipilimumab, CTLA4; pertuzumab, HER2; obinutuzumab, CD20; HER2; ofatumumab, CD20; ramucirumab, VEGFR2; siltuximab, IL6; pembrolizumab and nivolumab, PD-1; secukinumab, IL-17A; dinutuximab, GD2; alirocumab and evolocumab, PCSK9 (proprotein convertase subtilisin kexin type 9) inhibitor; idarucizumab, dabigatran; daratumumab, CD38; mepolizumab, IL-5; necitumumab, EGFR; elotuzumab, SLAMF7; obiltoxaximab, bacillus anthracis; ixekizumab, IL17A; reslizumab, IL-5; atezolizumab, PD-L1; daclizumab, CD25; adalimumab, TNF; ustekinumab, IL-2 and IL-23; olaratumab, PDGFR-α; bezlotoxumab, clostridium difficile toxin B
18.3.1 Mechanisms of Action of Tumor-Targeting Antibodies
Monoclonal antibodies have multiple intrinsic properties which can be exploited against cancer cells. Antitumor monoclonal antibodies can have a direct cytotoxic effect on cancer cells by directly inducing tumor cell death upon ligation to the tumor-specific antigen expressed on their outer membrane. Monoclonal antibodies can also be selected for their antagonist properties on tumor growth factors, binding either the ligands or blocking their cognate receptor (Fig. 18.4). Also, when monoclonal antibodies are opsonizing the surface of cancer cells, their distal part (the Fc domain) can trigger the destruction of the targeted cell upon engagement of the host immune effector mechanisms. First, they can activate the enzymes of the complement system which can directly attack the membrane of cancer cells or recruit immune effector cells via the complement receptor (CDC or complement-dependent cytotoxicity). Second, they may also activate cytotoxic innate immune effectors such as NK (natural killer) cells via the activation of their Fc gamma receptors (ADCC or antibody-dependent cellular cytotoxicity) (Fig. 18.4). Finally, upon opsonization of the targeted cells, the Fc domain of monoclonal antibodies can induce phagocytosis of cancer cells via the activation of Fc gamma receptors (FcγR) expressed on monocytes/macrophages (ADCP or antibody-dependent cellular phagocytosis) [9].
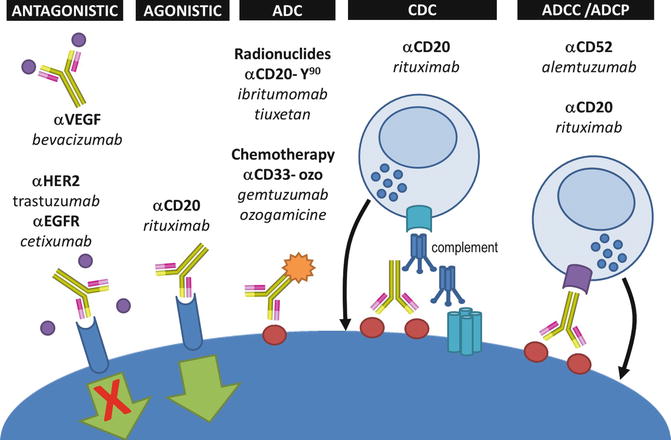
Fig. 18.4
Mechanisms of action of tumor-targeted monoclonal antibodies. Antibodies can be designed to either block a receptor or its ligand (antagonistic), or they can have intrinsic agonistic activities by directly stimulating receptors (e.g., pro-apoptotic signal). Tumor-targeted antibodies can be bound to either radioactive or chemotherapies to deliver the cytotoxic effect directly into the tumor bed (ADC antibody drug conjugate). The engineering of the Fc domains of antibodies can also enhance their ability to trigger complement-derived cytotoxicity (CDC), antibody-derived cell cytotoxicity (ADCC), or antibody-derived phagocytosis (ADCP)
18.3.2 Engineering of Tumor-Targeting Monoclonal Antibodies
The role of Fc-mediated cancer cell death has been demonstrated in retrospective clinical studies showing the prognostic value of FcγR polymorphisms in B-cell lymphoma patients treated with anti-CD20 monoclonal antibodies [10] and in children with neuroblastoma treated with anti-GD2 monoclonal antibodies [11]. Therefore, most tumor-targeted antibodies have been designed with an IgG1 isotype known to have a good affinity for Fc gamma receptors, notably CD16a (also known as FcγRIIIa), and an ability to activate natural killer cells (NK cells) to perform antibody-dependent cellular cytotoxicity (ADCC). Although the rationale for using IgG1 has been mainly built on blood NK cells, recent data suggest that IgG2 can also perform ADCC via FcγR expressing myeloid cells [12].
Within the CH2 region of antibodies Fc domain, amino acids (notably Asn297) can undergo glycosylation. The type of glycosylation of antibodies has an impact on the antibody FcγR affinity and its ability to trigger antibody-dependent cellular cytotoxicity [13]. The modification of monoclonal antibodies glycosylation status can now be engineered in order to enhance ADCC (Fig. 18.5). For example, obinutuzumab, a defucosylated anti-CD20 monoclonal antibody with enhanced ADCC potency in comparison to rituximab, has been approved by the FDA in 2013 for the treatment of chronic lymphocytic leukemia.
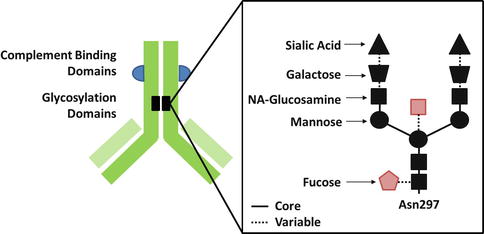
Fig. 18.5
 Peptide-Based Therapeutic Cancer Vaccines
Peptide-Based Therapeutic Cancer Vaccines
 The Human Tumor Microenvironment
The Human Tumor Microenvironment
 PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
 Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
 Immune Checkpoint Inhibition in Lung Cancer
Immune Checkpoint Inhibition in Lung Cancer
 Immune Monitoring of Blood and Tumor Microenvironment
Immune Monitoring of Blood and Tumor Microenvironment




Glycosylation status of monoclonal antibodies. Defucosylation or ablation of bisecting N-acetyl glucosamine (in red) enhances the affinity of monoclonal antibodies for Fc receptors and their ability to trigger antibody-derived cell cytotoxicity (ADCC)
Related posts:
Peptide-Based Therapeutic Cancer Vaccines
The Human Tumor Microenvironment
PD1 Checkpoint Blockade in Melanoma: From Monotherapy to Combination Therapies
Cancer Immunosurveillance by Natural Killer Cells and Other Innate Lymphoid Cells
Immune Checkpoint Inhibition in Lung Cancer
Immune Monitoring of Blood and Tumor Microenvironment
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
