Fig. 5.1
Focal nodular hyperplasia. (a) Mass magin is clear and the fibrous scar can be seen in the center of the lesion; (b) The color of the mass is similar with the surrounding livers, nodular appearance; (c) multi-mininodular lesions with unconspicuous scar or nodular appearance
5.1.1.4 Microscopic Features
There is clear boundary between the tumor and surrounding liver tissue, with disappearance of the normal structure of hepatic lobules in the tumor lesions. FNH can be classified into classical FNH and nonclassical FNH according to microscopic features.
Classic FNH , accounted for more than 90% in FNH [7], consists of proliferated liver cells, bile duct, fibrous tissue, and vascular malformation, with varying amounts of inflammatory cells (Fig. 5.2) and a nodular performance (Fig. 5.2a). In the center of the lesions are scars formed by fibrous hyperplasia and vascular malformation. The vascular malformation has abnormal vascular intimal thickening due to fibrosis, uneven vessel wall thickness, and poor internal elastic membrane formation (Fig. 5.2b). The central scar tissue protrudes fibers to segment the entire lesion in nodular appearance, and it is very difficult to distinguish the obvious heperplasia of the fibers from liver cirrhosis (Fig. 5.2a, b), which is the reason for the previous term of “focal hepatic cirrhosis” for FNH . The liver cells in the lesion are similar to normal hepatic cells without atypia in funicular arrangement within two layers. Hyperplasia of small bile ducts is arranged in clusters in the boundary of fibrous tissue and liver cells, some of which are in poor tubular formation (Fig. 5.2c). And varying amounts of lymphocytes infiltrated the interstitial fibrous tissue. A proportion of liver cells in the lesion have changes such as fatty degeneration, and extramedullary hematopoiesis occurs in some interstitial tissues, and FNH complicated by metastasis adenocarcinoma has also been reported (Fig. 5.2d) [7]. The surrounding liver tissue of FNH is non-cirrhotic, though some may have fatty degeneration or be accompanied by HCC or cholangiocarcinoma.
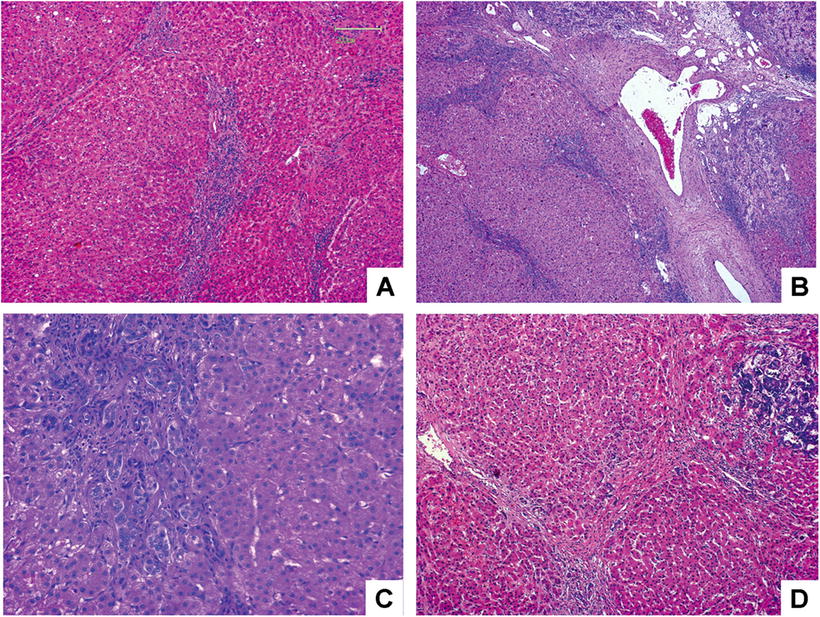
Fig. 5.2
FNH . (a) Hyperplasia fibrous segment the entire lesion in nodular appearance; (b) the center of the lesions is scars formed by hyperplasia fibrous and vascular malformation; (c) hyperplasia of small bile ducts, and inflammatory cells infiltrated; (d) FNH combined with adenocarcinoma metastatic
Nonclassical FNH is rare and considered to be variants of FNH . Previous classification of nonclassical FNH includes telangiectatic FNH , mixed hyperplastic and adenomatous FNH , and atypical FNH . Telangiectatic FNH has been reported to be monoclonal and is similar to hepatocellular adenoma in molecular variation, with significantly different clinical manifestation from classic FNH , such as frequent rupture and bleeding. Thus, it has been classified as a special subtype of hepatocellular adenoma [9]. Few reports of other two subtypes are found in the literature, and there are no widely adopted diagnostic criteria for them.
5.1.1.5 Immunohistochemistry
Catenin pathway activation leads to higher expression of downstream glutamine synthetase (GS) in FNH . The most important immunohistochemical markers are GS, CD34, CK7, and Ki-67 (Fig. 5.3). GS is expressed in hepatic cells around hepatic veins in a map-like distribution, and CD34 is mostly expressed in liver sinusoids diffusedly with focal distribution in some cases, while CK7 can be used to detect and outline the proliferated small bile ducts and Ki-67 suggests low proliferation index of FNH , mostly <1%. Other markers expressed in HCC are not detected in FNH , such as glypican-3, HSP70, and AFP.
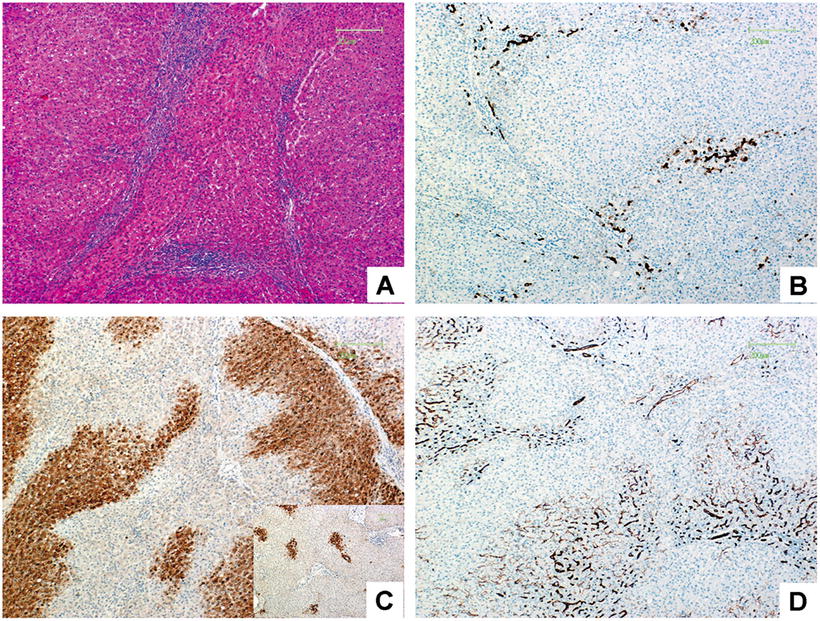
Fig. 5.3
Classic FNH . (a) The liver tissue was segment into nodules by fibrous, small bile hyperplasia ducts and inflammatory cells infiltrated in fibrous scar; (b) CK7 staining showed hyperplasia of small bile ducts in the boundary of fibrous tissue and liver cells; (c) GS staining showed positive cells distributed like a map, while the positive cells in the normal liver tissue were located in the periphery of the small hepatic vein showed as the right-down figure; (d) CD34 staining showed rich blood sinus distributed in focal or diffused
5.1.1.6 Differential Diagnosis
FNH should be differentiated from hepatocellular adenoma and HCC. Hepatocellular adenoma is a hepatocellular clonal proliferated lesion without bile duct and occurs mainly in young adults aged 30–40 years old with no history of hepatitis or liver cirrhosis, which is easily differentiated from FNH . However, in inflammatory hepatocellular adenoma with reaction of bile duct, GS is a helpful indicator for differentiation according to its map-like pattern in FNH , diffused distribution in hepatocellular adenoma with β-catenin mutation, and negative result in other types of hepatocellular adenoma . HCC occurs mainly in older men with the history of chronic hepatitis and liver cirrhosis, containing atypical tumor cells with increased cell karyoplasmic ratio, thickened liver plates often more than three layers, and positive results of immunohistochemistry detection including alpha-fetoprotein (AFP), glypian-3 staining, and HSP70.
5.1.1.7 Treatment and Prognosis
In a follow-up study of 30 patients with 34 FNH lesions by ultrasound examination every 3–6 months over an average period of 42 months, the results showed that the lesions remained stable in 24 cases (70.6%), deteriorated in 1 case (2.9%), and improved or disappeared in 9 cases (26.5%). In the six patients with disappeared lesions, older age and longer period were independent factors, and their average period for the disappearance of lesions was 59 ± 30 months [12]. FNH presents no malignant transformation but may occasionally rupture and bleed [13]. For asymptomatic small lesions, regular follow-up is recommended, and surgical treatment should be applied to those with large tumor masses, significant symptoms, undefined diagnosis, or rapid tumor growth during clinical observation [6].
5.1.2 Nodular Regenerative Hyperplasia
Wen-Ming Cong4 and Xin-Yuan Lu4
(4)
Department of Pathology, Eastern Hepatobiliary Surgery Hospital,, Second Military Medical University,, Shanghai,, China
5.1.2.1 Pathogenesis and Mechanism
Nodular regenerative hyperplasia (NRH) was first described as a “miliary liver adenomatosis” by Ranstrom in 1953 and named as NRH by Steiner in 1959. The mechanism involves hepatic portal vein blood flow disorder caused by various factors, such as portal vein tumor thrombus derived from primary or metastatic hepatic tumors, hematopathy, blood disease, the toxicity of chemotherapy drugs (including thiopurine drugs, anti-retroviral drugs [14, 15]), connective tissue disease, primary biliary cirrhosis (PBC), primary sclerosing cholangitis (PSC), hepatopulmonary syndrome (HPS) after liver transplantation, Budd-Chiari syndrome, immunological diseases (Felty syndrome, rheumatoid arthritis, splenomegaly, and leukopenia), myeloproliferative disease, tuberculosis, lymphoma , macroglobulinemia, polycythemia, renal transplantation, and HIV infection [16]. The common characteristics of the above diseases are endothelial inflammation and microvascular thrombosis inside the portal vein, resulting in deposition of red blood cells in Disse spaces of liver sinusoids and small branches of portal vein, causing obstruction or reduction in number, leading to sinusoidal obstructive syndrome (SOS), noncirrhotic intrahepatic portal hypertension (NCIPH), and further ischemic atrophy of the involved liver parenchyma and secondary reactive hyperplasia related to increasing blood flow of other small branches of portal vein, thus forming diffuse nodular hyperplasia without fibrous septa [17].
5.1.2.2 Clinical Features
NRH is the second most common cause of non-cirrhotic portal hypertension, with hepatosplenomegaly, portal hypertension, and other complications, such as esophageal varices and ascites, in half or more patients. NRH occurs mostly in adults. Jinjing Liu et al. reported 22 cases of NRH with an average age of 43 years old (22–70 years old) [18]. In the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, eight cases of NRH have been diagnosed since 1982, with male to female ratio of 7:1, aged 29–55 years old with an average of 44 years old. Lin Fang et al. reported a male patient of 34 years old who was admitted because of 1 year’s fatigue and abdominal distension in 2011 [19]. And the color Doppler ultrasound suggested liver cirrhosis, splenomegaly, and portal hypertension, and liver MRI demonstrated multiple small nodules in the liver, compression and attenuation of the hepatic vein, and enlargement of the spleen and splenic vein. Liver puncture biopsy showed complete structure of liver tissue, crowded liver plates, nodular hyperplasia of liver cells with no fibrous enclosure, and atrophy of surrounding liver cells due to compression. Albuquerque [20] reported a case of familial NRH in 2013 in which the mother and daughter both had NRH with non-cirrhotic portal hypertension and similar complications including variceal hemorrhage and hepatopulmonary syndrome.
5.1.2.3 Gross Features
Lesions are diffuse nodules in the liver with a granular envelope, similar to small cirrhotic nodules, with brown or yellowish brown section lighter than the color of the surrounding normal liver tissue. The common size of the lesions is within 5 mm in diameter, usually 1–3 mm; thus, they were called micronodular transformation (Fig. 5.4). In a few cases, the nodular size reaches 5 cm due to nodular fusion.
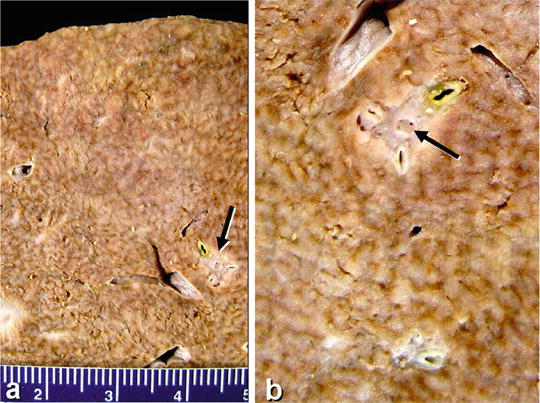
Fig. 5.4
Nodular regenerative hyperplasia. Small nodules ≤ 3 cm in diameter, the portal vein tumor thrombus in the liver metastasis of breast cancer is indicated by the arrow (Cite from Turk AT)
5.1.2.4 Microscopic Features
Liver lobules with NRH nodular hyperplasia contain diffused nodules instead of normal tissue with disorderly structure and similar size compared to normal tissue. The proliferated liver cells show no atypia and are arranged into 1–2 layers of liver plates which go in various directions and interleaved with each other instead of converging into central vein. Hepatocellular boundary is observed between the nodules with no fibrous septa (Fig. 5.5), and reticular staining shows inter-nodular atrophy of the liver tissue and collapse of reticular scaffold. Usually, NRH nodules distribute around portal vein branches, especially in the portal area. In almost all cases, small branches of portal vein less than 0.05 cm in diameter contain general stenosis and occlusive lesions which are rarely observed in punctured tissues. Hepatic cells are enlarged or weakly dyed with loose and translucent cytoplasm, due to varying amounts of intracellular glycogen and fatty vacuoles, and internodular hepatic cells show atrophy with decreased volumes and red-dyed cytoplasm due to compression, thus forming the specific structure of bright region and dark region. Lysozyme staining shows the endothelial cells in the hepatic sinusoids and Kupffer cells inside the nodules. Besides, each NRH nodule often involves only one portal area and is also known as single acinar regenerative nodules.
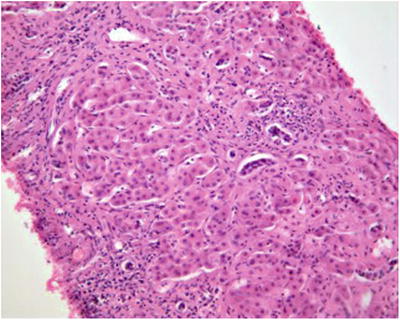
Fig. 5.5
Liver biopsy tissue showed hepatocellular hyperplasia nodules
Based on the above, the diagnostic criteria of NRH include: ➀ diffused hepatocellular hyperplasia nodules in the whole liver, ➁ intranodular hyperplasia and atrophy of liver cells, ➂ no fibrous envelope around the regenerative nodule, and ➃ no or rare inflammation in the portal area.
5.1.2.5 Differential Diagnosis
NRH are similar to small nodular cirrhosis both clinically and in gross appearance. In NRH, the nodules are soft, and the internodular boundary is unclear with gradual transition between intra- and extra-nodular liver cells. Non-internodular fibrous septa are the main histological feature to differentiate NRH from cirrhosis regenerative nodules. Masson trichrome staining shows no fibrous envelope around the nodules. NRH should also be differentiated from hepatic focal nodular hyperplasia , hepatocellular adenoma , congenital hepatic fibrosis (CHF), and HCC (Table 5.1). Liver puncture biopsy can be used to diagnose but with much difficulty, and an open wedge liver biopsy is alternative when necessary [21].
Table 5.1
Differential diagnosis of common nodular regenerative hyperplasias
Diseases | Pathological features |
|---|---|
Nodular regenerative hyperplasia | Multiple lesions in the liver, consistent in size, without internodular fibrous septa, with stenosis, occlusion, or malformation/deviation of branches of the portal vein |
Regenerative nodules in hepatic cirrhosis | A single lesion or multiple lesions with obvious inflammation and necrosis, various sizes, surrounded by inflammatory fibrous tissue |
Focal nodular hyperplasia | A single nodule with stellate scar in the center consisting of multi-branched fibrous tissue which is rich in thick-walled vessels, and the vessel wall contains fibromuscular hyperplasia and concentric or eccentric stenosis in the vessels. |
Hepatocellular adenoma | A single tumor or a few large tumor nodules, without portal area involvement |
5.1.2.6 Treatment and Prognosis
NRH is a benign lesion and should receive symptomatic treatment due to the increase in the size and number of lesions, and the major complications are portal hypertension and late-term liver failure. The prognosis of NRH is generally good if the portal hypertension is well treated. Radomski et al. (2000) reported four cases of liver transplantation in NRH patients, who survived with normal hepatic function during the 2–4 years’ follow-up. Alhosh et al. [22] reported a case of a 3-year-old boy who was diagnosed as NRH and received the first liver transplantation, and he received a second liver transplantation 2 years later due to hepatopulmonary syndrome with good recovery, normal hepatic function, improved hepatopulmonary syndrome, and normal oxygen saturation both at rest and in activity [22]. There is no evidence of canceration of NRH; however, Sood et al. [21] reported HCC in a 44-year-old woman with Turner syndrome, and the relevance of HCC and Tuner syndrome should be further investigated [23].
5.1.3 Partial Nodular Transformation
5.1.3.1 Pathogenesis and Mechanism
Partial nodular transformation (PNT) was first named by Sherlock and colleagues in 1966, though similar lesions were first reported early in 1090. PNT is an independent lesion, and more than ten cases have been reported, while some people regard it as a variant of NRH. PNT is characterized by large nonfibrous hyperplastic nodules formed by hepatocellular in nonfibrous or non-cirrhotic liver, which is also called non-cirrhotic nodule, primarily distributed along larger intrahepatic portal vein branches or portal tracts in periportal area. The pathogenesis of PNT is considered to be obstruction of the vessels in the hepatic hilar region or major branches of intrahepatic portal veins or hepatic veins, such as thrombosis in Felty syndrome (characterized by rheumatoid arthritis, with splenomegaly and reduction of white blood cells), which facilitates the communication of the obstructed vessels with the main portal veins in the portal area, resulting in regional hypoperfusion leading to atrophy of the liver parenchyma, and portal hyperperfusion to form fusion of multiple nodules. Hoso et al. (1996) reported a case of multiple PNT surrounding the intrahepatic tumor thrombus in the main portal vein, and the largest nodule was 8 cm in diameter without a fibrous envelope. PNT caused by secondary thrombosis were also reported in literature, due to occlusion of portal venous branches and portal cirrhosis in patients with idiopathic portal hypertension (IPH). Wanless et al. (1985) reported one case of a 29-year-old male with patent ductus venous, 15 mm in length and 15 mm in diameter. In this case, a portion of the portal venous blood was directly drained into the inferior vena cava via the umbilical vein, and several fistulae communicated the patent ductus venous with left hepatic vein and inferior vena cava, leading to dysplasia of intrahepatic portal venous vasculature and hyperplasia of partial liver parenchyma, which contributed to the formation of two 4 cm-in-diameter PNT nodules.
5.1.3.2 Clinical Features
Patients’ clinical manifestation is characterized by presinusoidal portal hypertension.
5.1.3.3 Gross Features
PNT appears to be multiple nodules in varying sizes, with diameter of 0.3–4 cm, and is also called macronodular transformation.
5.1.3.4 Microscopic Features
The hyperplastic nodules distribute surrounding main branches of portal veins with blood or tumor thrombus in them. The nodules consist of proliferated liver cells and show the arrangement of 2–3 layers of liver plates via reticular fiber staining, with visible capillary bile duct and bile thrombi. Internodular region is composed of atrophic liver cells, without the formation of fibrous septum in Masson staining. Though PNT and NRH are similar histologically, PNT nodules tend to form larger nodules which are often several centimeters in diameter and are located mainly in the hepatic hilar region or along main branches of portal vein, while NRH are distributed diffusedly.
Supplementary: Idiopathic Portal Hypertension
Idiopathic portal hypertension (IPH) is classified as a benign nodular hepatocellular lesion, as well as NRH and FNH , and is related to liver hemodynamic disorder, characterized by increased flow resistance of presinusoidal portal veins and portal hypertension. IPH commonly appears in diffused lesions, due to chronic ischemia leading to hepatic parenchymal atrophy and nodular regeneration. However, nodular hyperplasia has been discovered in many cases. In addition, IPH, also known as non-cirrhotic portal fibrosis (NCPF) [24], shows undefined boundary without fibrous septa under the microscope. The portal fibrous tissue and bile ducts proliferate and stretch out into the hepatic parenchyma, with mild edema and infiltration of lymphocytes, and there is no interface inflammation or necrosis of hepatocytes. And the liver tissue is surrounded by incomplete fibrous envelope forming nodules with occlusion in small branches of portal veins and cirrhosis of central vein. IPH-related hepatic nodular lesions are benign disease with rare report of canceration.
5.1.4 Precancerous Lesions
Wen-Ming Cong5, Long-Hai Feng5 and Guang-Zhi Jin5
(5)
Department of Pathology, Eastern Hepatobiliary Surgery Hospital,, Second Military Medical University,, Shanghai,, China
Among precancerous lesions , dysplastic foci (DF) and dysplastic nodule are of the most importance [25].
5.1.4.1 Dysplastic Foci
Pathogenesis and Mechanism
Dysplastic foci (DF ), first proposed in the international consensus of terminology of hepatic nodular lesions by International Working Group (IWP) of World Gastroenterology Organisation in 1995, were defined as small liver cell dysplasia (LCD) with a diameter <1 mm which cannot be identified by radiological imaging or with the naked eye [26, 27]. DF are often multiple lesions in patients with chronic hepatitis, especially those with liver cirrhosis. LCD was described by British pathologist Anthony in 1973 for the first time, and he suggested that LCD was a precancerous lesion of HCC. Later Japanese researchers, Watanabe et al., classified LCD into two major types, small-cell dysplasia and large-cell dysplasia, in 1983, which are now known as small cell change and large cell change, in which DF is consisted mainly of small cell change or large cell change.
Microscopic Features
- 1.
Small cell change. Small liver cells with decrease in size and increase in nucleocytoplasmic ratio, and the cytoplasm is basophilic. The nucleus is mildly pleomorphic and atypical, darker in color and crowded-like. In some LCD, multinuclei is the major characteristic. The proliferative activity of small cell change is higher than that of the surrounding liver tissue, similar to the performance of early HCC (Fig. 5.6a). The risk of canceration of small cell change-dominated DF with rapid proliferation is generally considered higher [28].
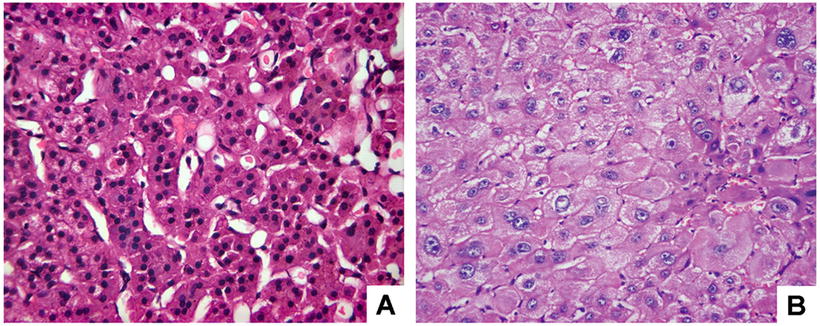
Fig. 5.6
Liver cell dysplasia. (a) Small cell change: hepatocyte was decreased in cytoplasm and size, increased in nucleocytoplasmic ratio, and occasional nuclear division; (b) large cell change: hepatocyte was increased in size and their nuclei, nucleocytoplasmic ratio was almost normal, and multinucleate hepatocyte was observed
- 2.
Large cell type. Hepatocytes in this type increase in size in proportion to their nuclei, resulting in normal nucleocytoplasmic ratio, with normal cellular density and cytoplasmic staining, while nuclear envelopes thicken and shrink, and atypia of hepatocytes with pleomorphic and dark-stained nuclei is commonly observed (Fig. 5.6b). Large cell change was previously considered as the feature of LCD by Anthony; however, whether LCD is a precancerous lesion is still in discussion, and WHO classification has not defined the property of LCD. Based on the literature, LCD with a history of HBV-/HCV-related liver cirrhosis is an important precancerous lesion supported by most researched. Since 1988, we have reported the results that the DNA ploidy and quantitative features of nuclear morphology are between that of normal liver cells and HCC cells, in consistent with the basic feature of precancerous lesion, suggesting LCD is atypia of cell population in various precancerous stages [29, 30].
Clinicopathological Significance
The occurrence of DF and LCD is relevant to the risk of HCC. Studies have shown that the incidences of LCD in the tissue of simple liver cirrhosis and pericarcinomatous hepatic cirrhosis were 6.9% and 64.5%, respectively, and the risk ratio of HCC in non-liver cirrhosis male patients with or without LCD was 13:1. The average age of liver cirrhosis patients without LCD was younger by 4–10 years than that of patients with LCD, while the average age of liver cirrhosis patients was younger by 3–6 years than patients with HCC, based on which we deduce that it would take 7–16 years for the development of liver cirrhosis to LCD to HCC. Thus, if crowded or clustered LCD is found in the pericarcinomatous liver cirrhosis tissue, it is necessary to pay more attention to them and follow up for a longer time. And it has been calculated that the risk of HCC derived from small cell change and large cell change (odds ratio, OR) was 6.33 and 3.88, respectively, suggesting that small cell change is more likely to cancerate.
5.1.4.2 Dysplastic Nodule
Pathogenesis and Mechanism
Dysplastic nodule (DN) is a nodular lesion, different from the surrounding liver tissue in morphology such as size, color, texture, section, etc., and was previously named adenomatous hyperplasia. It primarily occurs in patients with HBV-/HCV-related chronic hepatitis or liver cirrhosis, as well as chronic hepatic injuries, such as chronic alcoholic liver cirrhosis. DN is classified into low-grade dysplastic nodule (LGDN) and high-grade dysplastic nodule (HGDN) according to the degree of atypia of hepatocytes inside the lesion, and the common characteristic is the clonal proliferation of cell populations, including accumulation of intracellular iron and copper, fatty, and hyaline degeneration of hepatocytes, and eosinophilic degeneration, all of which do not exist in the surrounding liver tissue.
Clinical Features
Up to now, a total of 135 cases of DN have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, since 2005, with 57 cases of simple DN and 57.8% cases complicated by other diseases such as HCC, intrahepatic cholangiocarcinoma , hepatocellular adenoma , and cavernous hemangioma . Among them, more patients are male with a male to female ratio of 5.83:1, and the onset age ranged 27–75 years with an average of 54.6 years, and 95.7% of the patients had a history of viral hepatitis or cirrhosis, and three patients had chronic schistosomiasis liver disease. Most patients had no obvious clinical manifestation and were found to have liver lesions on examination. The main clinical manifestations include upper abdominal discomfort, abdominal pain, and other nonspecific digestive system symptoms. Some of the patients have portal hypertension due to liver cirrhosis or corresponding clinical symptoms and signs when complicated by HCC. In our hospital, serum AFP in LGDN and HGDN patients ranged 0–1210 U/L and 1.5–355.9 U/L, respectively. Ultrasound B examination often presents an inhomogenously hypoechoic area. CT images demonstrate hypodense area with irregular arterial enhancement and slightly lower density than the surrounding normal liver tissue in the portal and late phases of enhanced CT scanning. MRI shows hypointensity, isointensity, or hyperintensity on T1WI and hyperintensity on T2WI, and enhanced MRI demonstrates the similar change as CT images that irregular enhancement of the lesion is observable with decreased signals in venous phase and balance phase.
Gross Features
DN occurs more in the right lobe of the liver, with a single lesion or multiple lesions, or even more than ten nodules in some cases, among which the degrees of atrophy may be different and some nodules may have cancerated. The average diameters of LGDN and HGDN lesions are 1.85 (0.5–4.1) cm and 2.56 (0.6–5.2) cm. DN is gray-white, obviously different from the color of the surrounding liver tissue. The cut surface shows lesion bulge out of the surrounding liver parenchyma, gray-yellow or gray-white in color, with a small amount of hemorrhage and necrosis, with a complete thin envelope or an ill-defined boundary (Fig. 5.7).
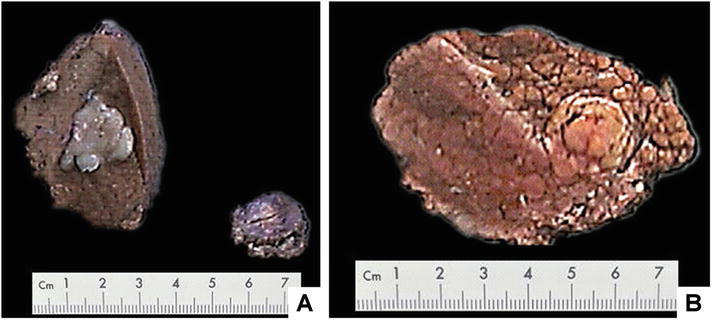
Fig. 5.7
Dysplastic nodule . (a) Small hepatocellular carcinoma in liver specimen, small nodule at right was LGDN resected simultaneously; (b) HGDN , the nodule had a roughly clear boundary, and no fibrous envelope
Microscopic Features
The histological criteria of DN were proposed by International Consensus Group for Hepatocellular Neoplasia (ICGHN) in 2009 [31].
- 1.
LGDN . The nodules have an ill-defined boundary, and there is a fibrous septum to separate it from the surrounding liver cirrhosis which is a pseudoenvelope. Large cell change is visible in some lesions while small cell change is rare. The cells increase in density with uniform arrangement. Despite the existence of large cell change, there is no atypia or pseudoglandular tubes, with insignificant broadening of hepatic trabecula, mild capillarization in hepatic sinusoid, and few isolated arteries without concomitant bile duct (Fig. 5.8). LGDN has a blood supply of portal vein and is difficult to distinguish it from large regenerative nodules (LRN) morphologically, and ICGHN has grouped LRN into LGDN [25].
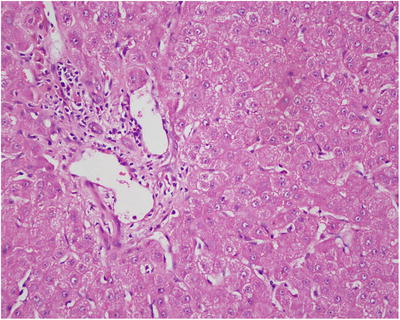
Fig. 5.8
LGDN portal structure was basically integral
- 2.
HGDN . The nodules in HGDN have a well- or ill-defined boundary and have no fibrous envelope, with mild atypia in cell morphology and structure and cannot be diagnosed as HCC. The cell density increases (two times the surrounding liver tissue), and the liver cells are arranged in irregular trabecula, with pseudoglandular tubes, with significant broadening of hepatic trabecula (Fig. 5.9). Small cell change is more commonly seen, with hyaline degeneration or fatty degeneration of liver cells. Different from HCC, there are a few portal structures in HGDN , with increased isolated arteries without concomitant bile duct (Fig. 5.10). And reticular structure disappears with ductular reaction visible suggesting the lesions may be nonmalignant with no stromal invation (nodular lesions with ill-defined boundary present invasion of the portal area and fibrous septa). In addition, we’ve found that HGDN has loss of heterozygosity (LOH) and instability of multiple genomic microsatellites which need further investigation for application in diagnosis and differential diagnosis [32]. Thus, present criteria on the differential diagnosis of HGDN and well-differentiated sHCC need further improving via active practice to achieve a broader consensus [33–35].
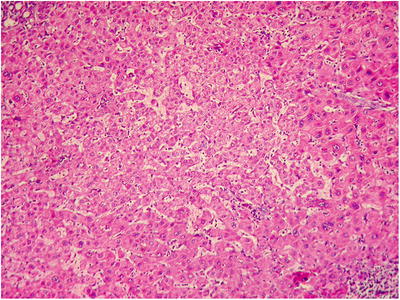
Fig. 5.9
HGDN trabecula was broadened in the central region of the lesion; atypical hyperplasia was obvious in the peripheral region
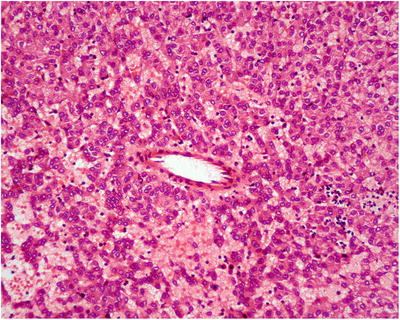
Fig. 5.10
Isolated arteries without concomitant bile duct
- 3.
Nodule in nodule. Well-differentiated carcinomous micronodules can be identified via imaging or by the naked eye, with stromal invasion, which indicates invasion of carcinomous cells into the intranodular portal area and fibrous septa, in DN , especially HGDN . The nodules grow aggressively and gradually replace the surrounding liver tissue with atypia (Fig. 5.11). We have diagnosed 30 cases of single HGDN in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, since 2005, 13 cases (43.3%) of which presented nodule in nodule [36].
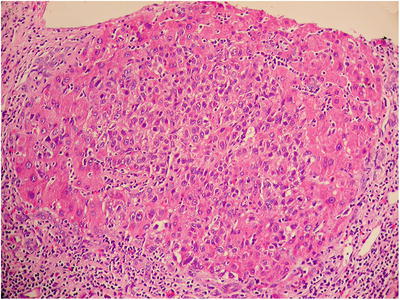
Fig. 5.11
HGDN , nodule in nodule of cancerization in the center of HGDN , expansive growth and transition with peripheral hepatic cell plate
Immunohistochemistry
CK7 and CK19 are important markers to define ductular reaction and invasion. Nodules in HGDN present bile duct stain in the portal area, with increased Ki-67 index, and CD34 staining reveals the level of capillarization in hepatic sinusoid, which is related to the degree of carcinomous differentiation [37]. And DN with positive GPC-3, especially HGDN with positive GPC-3, is a late phase of precancerous lesions [38]. Kim Kwang Shik et al. screened a group of differentially expressed protein in DN and HCC tissues using proteomic methods, and the results showed that the expression levels of ACY1 and SUOX decrease in LGDN , HGDN , and WD-SHCC successively, contrary to the expression situation of protein P62. And the combination of SUOX, CD34, and AKR1B10 tests is beneficial in the differential diagnosis of DNA and WD-SHCC [39].
Differential Diagnosis
- 1.
Focal nodular hyperplasia . The proliferated hepatocytes are well differentiated, cluster to form nodules, almost without a background of chronic hepatitis or liver cirrhosis. A typical fibrous scar is helpful in the diagnosis. And in nontypical scar cases, microvessels distributed along the fibrous scar in CD34 immunohistochemistry are an evidence for diagnosis.
- 2.
Hepatocellular adenoma . Hyaline degeneration and fatty degeneration may be observed in liver cells, sometimes with mild atypical hyperplasia and pseudoglandular tube. And diffuse, dialated, and thin-walled small vessels and vascular purpura-like changes are evidences for diagnosis.
- 3.
Well-differentiated sHCC . Cell density increases and intertrabecular space widens, with expansive growth and invasion of the stroma or adjacent liver tissue. CD34 staining shows significant increase of microvessel density.
Treatment and Prognosis
Cancerous property of DN is the most important feature for clinical pathology. Reports showed the risk ratio for canceration of HGDN , LGDN , and LRN is 16.8, 2.96, and 1, respectively. Surgical treatment, including radiofrequency ablation and surgical resection, is the preferred choice for HGDN with radical effects in precancerous lesions . Given the high risk of HGDN canceration, the postoperative patients in early or start-up phase of HCC should be closely followed up [40]. Tommaso et al. [25] proposed the risk assessment of developing HCC in DN patients (Table 5.2).
Table 5.2
Risk assessment of developing HCC in DN patients
Major indicators | Risk |
|---|---|
Diameter | |
<1 cm | Low |
>1.5 cm | High |
Nodular image in arterial phase | |
Low to moderate vasculature | Low |
Rich vasculature | High |
Enhanced MRI | |
Moderate or high density | Low |
Low density | High |
Follow-up imaging | |
Stable nodules | Low |
Enlargement of nodules | High |
With enhancement in arterial phase | Low |
Without enhancement in arterial phase | High |
Complication of HCC | |
With | Low |
Without | High |
Pathological feature | |
LGDN | Low |
HGDN | High |
5.1.5 Compensatory or Segmental Hyperplasia
The causes of compensatory or segmental hyperplasia of liver (CLH) include occlusion of portal vein or hepatic vein (Budd-Chiari syndrome), primary sclerosing cholangitis (PSC), stenosis or obstruction of the major bile duct (Allagille syndrome), and so forth, all of which lead to atrophy of involved liver parenchyma and partial or complete compensatory hyperplasia of other hepatic lobes, especially the caudate lobe, due to the its independent drainage in Budd-Chiari syndrome, while other lobes’ drainage is dependent on the hepatic vein. Imaging shows that CLH was a nodular lesion of several centimeters in diameter close to the liver capsule. Histologically, compensatory or segmental hyperplasia of the liver tissue remains the portal area and central vein, with normal hepatocytes and atrophic liver cells in the surrounding regions. It should be differentiated from hepatocellular adenoma and well-differentiated HCC.
5.1.6 Focal Fatty Change
5.1.6.1 Etiology and Mechanism
Focal fatty change (FFC), first reported by Simon in 1934 as focal fatty infiltration, is a benign tumor-like lesion without any invasive property. According to radiological performance, non-diffuse hepatic adipose infiltrations are classified into four types: focal, multifocal, hepatolobular or hepatic segmental, and liver focal fatty sparing. The causes of FFC have not been well defined, and reports abroad suggested that FFC may be related to alcoholic liver disease, steroids, diabetes, and abnormal increase of portal venous supply induced by various factors. We have reported several cases with the history of hepatitis B in some FFC patients, indicating the involvement of hepatitis in the pathogenesis of FFC.
5.1.6.2 Clinical Features
FFC is often discovered in adults. A total of 42 cases have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, with a male to female ratio of 1.1:1 and an average age of 48.7 years old (range 26–85 years old). The patients had no obvious symptoms, presented strong echoes in ultrasound B examination. In CT scanning, focal low-density areas with clear or undefined boundaries are detected, with relatively decreased enhancement compared to the normal liver tissue; thus, it is difficult to differentiate it from HCC, especially multifocal FFC which is like metastasis. In MRI T1 phase, FFC shows regional high signal with relatively weak enhancement; however, it is difficult to differentiate it from HCC in cases of heterogeneous fatty livers whose MRI is extremely similar to that of HCC. When clinical symptoms are inconsistent with imaging findings or there is contradictory diagnostic evidence, cautious decisions on therapeutic strategy should be made. And a liver puncture biopsy is the best choice to diagnose undefined lesions [41, 42].
5.1.6.3 Gross Features
The lesions may be solitary or multiple, nodular in some cases, or appear in combination with other hepatic diseases such as liver cirrhosis and mostly seen in the right lobe of the liver, colored light yellow or dark red, 3–5 cm in diameter or larger than 10 cm in diameter in some cases, as tender as the liver but with no capsule, and one can distinguish it from the liver due to the difference in color and luster between them (Fig. 5.12). Although the imaging study suggests intrahepatic nodules, they can rarely be identified by the naked eye; thus, a guidance of ultrasound B should be introduced to detect the lesions.
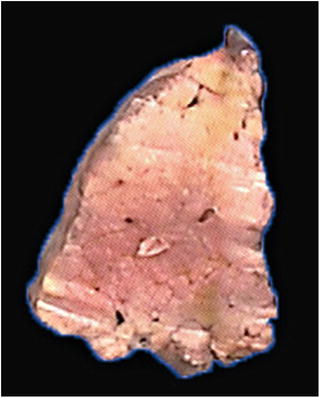
Fig. 5.12
Focal fatty change in the lesion area is located in the upper part of the specimen, light yellow, without coating
5.1.6.4 Microscopic Features
The liver cells of fatty change in the lesion is focal or in the form of small patches, with macrovesicular lipid droplets in most of the cells. Lesions are located around the central vein or form fusion lesions, with no atypia of liver cells. Mallory bodies can be observed in the cytoplasm of hepatic cells in patients of alcoholic hepatic disease with no reconstruction of hepatic lobules, and portal area is visible with expansion and congestion of portal venous branches (Fig. 5.13). The degree of fatty change in the surrounding areas shows a gradient decrease, and liver cirrhosis occurs in patients with severe or long-term disease.
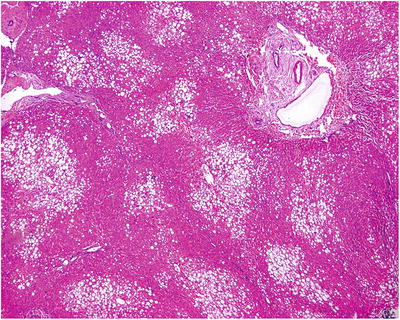
Fig. 5.13
Focal fatty change. Focal hepatic steatosis in the lesion area, distribution along the central vein
5.1.6.5 Differential Diagnosis
The clinical pathological significance of FFC lies in the differential diagnosis from tumors. Liver puncture biopsy results show a transition region between FFC and normal liver tissue, which is beneficial for diagnosis. In severe FFC, it should be differentiated from lipoma and well-differentiated liposarcoma. Lipoma has the same immunohistochemistry profile of liver cells and should be identified from lipid-rich well-differentiated HCC; well-differentiated liposarcoma has significant atypia, and lipid-rich angioleiomyolipoma contains a small amount of epithelioid cells surrounding adipocytes, which is positive in immunohistochemical HMB45 staining, and proliferation of small vessels different from the central vein. The key point to different FFC from hepatic adipose infiltration is the distribution of fatty change, as the former is characterized by fatty change surrounding the central vein and macrovesicular lipid droplets in most of the cells, and the latter diffuse or focal fatty change.
5.1.6.6 Treatment and Prognosis
FFC is a benign disease and should receive symptomatic treatment instead of any special treatment.
5.1.7 Accessory Lobe
Wen-Ming Cong6 and Xin-Yuan Lu6
(6)
Department of Pathology, Eastern Hepatobiliary Surgery Hospital,, Second Military Medical University,, Shanghai,, China
Accessory lobe is a type of congenital liver anatomic variations, consisting of normal liver tissue, with an incidence rate of 0.44%, and is often discovered in open or laparoscopic surgeries or autopsy, either connected to liver tissue or located in areas, such as the posterior or superior surface of the right lobe of the liver, gallbladder bed [43], or even thoracic cavity in the form of herniation in some reports [44]. Accessory lobe contains independent systems of the hepatic artery, portal vein, bile duct, and hepatic vein and can be classified into four types, according to the direction of bile drainage and the condition of Glisson’s capsule: type I, drainage to intrahepatic bile duct of the normal liver; type II, drainage to the extrahepatic bile duct; and type III, drainage to the extrahepatic bile duct and shares hepatic envelope with the normal liver. Ruiz Hierro et al. [45] reported the resection of type I accessory lobe in the abdominal cavity in a 13-year-old child, which was 10 cm × 7 cm × 10 cm in size with focal nodular hyperplasia [45].
5.2 Bile Duct Tumor-Like Lesions
5.2.1 Biliary Hamartoma
Wen-Ming Cong7
(7)
Department of Pathology, Eastern Hepatobiliary Surgery Hospital,, Second Military Medical University,, Shanghai,, China
5.2.1.1 Pathogenesis and Mechanism
Biliary hamartoma (BH) was first reported by von Meyenburg in 1918 and is a combination of various components; thus, it is called von Meyenburg complex (VMC). It is also known as biliary microhamartoma (BMH) or multiple biliary hamartomas, due to the small and multiple lesions. BH is a rare focal developmental disease of the bile duct, accounting for 0.6–5.6% of autopsy cases and 0.6% of regular liver puncture biopsy cases. The pathogenesis of BH may involve the developmental malformation or abnormal reconstruction of bile duct plates (the precursor of the normal bile ducts) in embryo phase. In terms of embryology, intrahepatic bile duct plats derive from hepatic portal area to terminal development and reconstruction and are bilayer cylindrical in the embryo liver. The fetus bile duct plates in postnatal livers are known as bile duct malformation or hepatobiliary fibrous polycystic disease. The malformation is located in the primary branches of intrahepatic bile duct system, and the abnormal non-degenerative bile duct clusters in the liver are called Meyenburg clusters. Thus, BH is a congenital benign tumor-like lesion, as well as a disease in the category of fibrous polycystic lesions, including congenital hepatic fibrosis, Caroli disease, autosomal dominant polycystic liver disease (PLD), and congenital choledochal cyst (CCC) [46].
It has been reported that around 27% of BH patients are complicated with congenital hepatic fibrosis, 27% with FNH , and 20% with hepatic cysts; however, the incidence of BH is 41–100% in polycystic liver disease and approximately 100% in polycystic kidney disease. And 16.9% of the simple cystic liver cases are complicated by BH . Furthermore, multiple BH (>10 lesions) increases the risk of complication of FNH , hemangioma , congenital hepatic fibrosis, polycystic kidney, and cholangiocarcinoma . Though BH is proposed to be hereditary, no evidence has confirmed any BH -related autosomal dominant genetic variation.
5.2.1.2 Clinical Features
The majority of patients are middle-aged women with an average age of 42 years old, ranged 40–85 years old, and the average age of patients with giant BH is 69 years old. Clinical manifestations correlate to the size of the lesion, and most patients present no specific symptoms or signs. However, patients with giant BH have abdominal pain due to the compression of hepatic envelope, and a few patients present obstructive bile duct-related symptoms, including jaundice, cholangitis, fever, abdominal pain, or even portal hypertension, due to the mucous secretion and the complication of bile duct stones. B-mode ultrasonography showed small hypo- or hyperechoic cystic lesions or hyperechoic region between cystic lesions. CT demonstrates multiple small hypodense cystic lesions with no enhancement after administration of venous contrast agent. Dilatation of multiple intrahepatic bile ducts can be seen in liver parenchyma. Enhanced CT shows scattered round hypodense areas with dilatation of intrahepatic bile ducts, which is similar to the image of cholangiocarcinoma . In MRI, the lesions are round cystic masses with clear boundaries and thread-like contoured enhancement, which is hyposignal on T1-weighted images and hypersignal on T2-weighted images [47, 48]. And magnetic resonance cholangiopancreatography (MRCP) shows multiple hyperdensity lesions without communication between the cyst and the normal bile ducts.
5.2.1.3 Gross Features
The lesions in multiple BH are small, often 0.1–0.5 cm in diameter, and locate superficially under the Glisson’s capsule. They are diffuse, gray-white micronodules that are solid and tender in nature. In some cases, the nodules colored in yellow-white are distributed in the whole liver and appear like “stars.” Solitary BH lesions with clinical manifestation can be large. Martin et al. [47] reported 15 cases of BH , and the lesions were 2.7–21.6 cm in diameter with micro-lesions in the surrounding area, indicating that the giant lesions derived from micro-lesions via dilatation. On the section, BH lesions were small cystic cavities with smooth wall, and the cystic wall was 0.1–0.8 cm in thickness, and the cavities are multilocular with fibrous septa (Fig. 5.14), so they are also known as multicystic biliary hamartoma (MCBH).
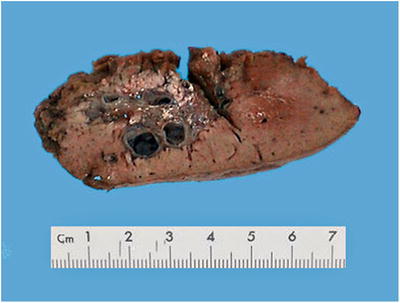
Fig. 5.14
Biliary hamartoma. The lesion was multilocular and no envelope
5.2.1.4 Microscopic Features
BH is a portal lesion, characterized by small bile duct plates closely located adjacent or in the portal area (Fig. 5.15) or in dense arrangement, with compression to the surrounding liver tissue (Fig. 5.16). The small bile ducts are immature in shape, with angled bend or branches, as well as various cystic dilatation and intraductular communication (Fig. 5.17), and contain bile thrombus, blood, or acidic proteinic secretion. The thin walls of bile ducts are lined with simple squamous epithelium or simple cuboidal epithelium, similar to the epithelium of bile ducts, with no nuclear atypia (Fig. 5.18). The cystic dilatated bile ducts open at the peritubular glands with or without mucous secretion (Fig. 5.19). The small bile ducts are embedded in dense fibrous stroma which is often with collagen degeneration or hyaline degeneration, and there is a fibrous ring around the lesions with clear boundary. It suggests canceration when the glands are in dense arrangement, expansive growth, or invasion of the surrounding liver tissue [47–50].
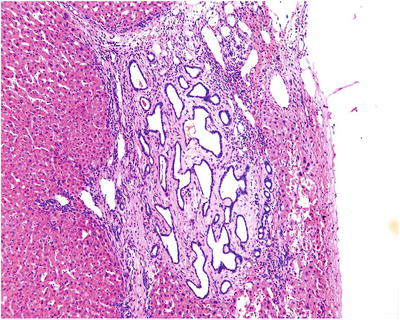
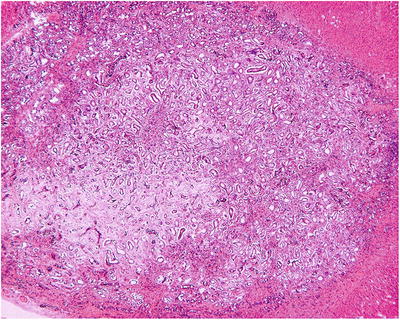
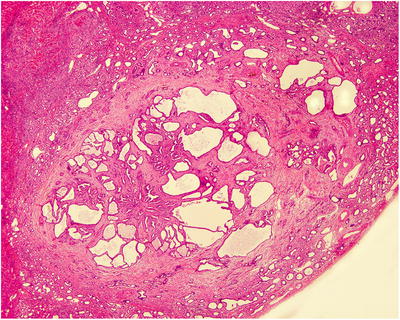
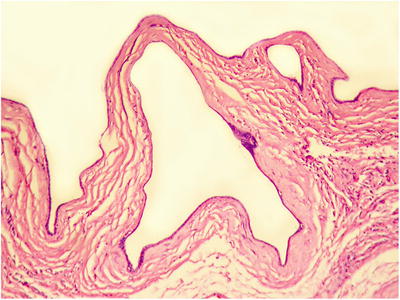
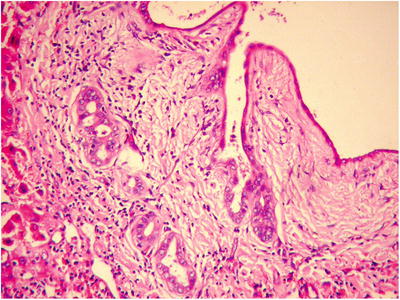
Fig. 5.15
The tumor is composed of cystic dilated small bile ducts
Fig. 5.16
Small bile ducts were dense hyperplasia with uniform shape and abundant fibrous stroma
Fig. 5.17
Cystic dilatation of the bile duct and intraductular communication
Fig. 5.18
Lumen lined with simple squamous epithelium or simple cuboidal epithelium with high dilatation
Fig. 5.19
Cystic dilatated bile ducts open at the peritubular glands
5.2.1.5 Immunohistochemistry
CK19 and CA19-9 are positive and CK20 is negative.
5.2.1.6 Differential Diagnosis
- 1.
Ciliated hepatic foregut cyst (CHFC). The cystic wall of CHFC has four layers, pseudostratified ciliated columnar epithelium, subepithelial connective tissue, smooth muscle, and outer layer of fibrous tissue, while the wall of MCBH doesn’t have ciliated epithelium.
- 2.
Congenital intrahepatic bile duct dilatation (Caroli disease (CD)). Caroli disease is characterized by segmental cystic or multicystic dilatation of intrahepatic bile ducts, while MCBH is derived from bile duct hamartoma or abnormal bile ducts, often combined with congenital hepatic fibrosis.
- 3.
Mucinous cystic neoplasm. It belongs to the category of biliary cystadenoma , with mucous epithelium-covered cystic wall underneath which is ovarian-like stroma. This is a tumor with potent canceration of biliary cystadenocarcinoma and should be carefully differentiated from MCBH.
Other diseases which should be differentiated from include bile duct adenoma , well-differentiated intrahepatic cholangiocarcinomas , metastatic adenocarcinoma, congenital hepatic fibrosis, and FNH , which are detailed in related sections.
5.2.1.7 Treatment and Prognosis
BH is a benign lesion, however, more than ten cases have been reported to develop into be complicated by intrahepatic cholangiocarcinoma , [51] especially in multiple BH which may cancerate via heperplasia, metaplasia, or dysplasia. Moreover, it has been observed the combination of BH and intrahepatic cholangiocarcinoma with a transitional region between them in histological examination. Thus, BH should be treated with surgery principally.
5.2.2 Simple Hepatic Cysts
5.2.2.1 Pathogenesis and Mechanism
According to the causes, mechanism, origin, histology, and relation with the bile duct, simple hepatic cysts (SHC) have various classifications. For example, hepatic cysts are divided into congenital and acquired groups. The former includes bile duct hamartoma, congenital hepatic fibrosis, solitary cyst, polycystic liver, Caroli disease, and common bile duct cyst. The latter includes parasitic, traumatic, and inflammatory tumors (such as cystadenoma , cystadenocarcinoma , teratoma ) and miscellaneous cysts (such as endometrial cyst), and traumatic and inflammatory cysts also belong to the category of pseudocysts.
SHC are congenital, non-hereditary intrahepatic cystic lesions, often referred to as the liver cyst, also known as solitary cyst of the liver. They are commonly seen and accounting for more than 95% of hepatic cysts. SHC are sporadic, with no familial heredity, and may derive from the persistent dilatation and fusion of the residual bile duct plates formed in embryonic phase or biliary hamartoma in which the epithelial hyperplasia of small bile ducts induces obstruction and accumulation of secretion, and SHC are also considered as retention cysts which are present at birth. Most SHC are not complicated by cysts in other organs. However, in the Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, a 46-year-old woman was found to have a 5.3 cm × 4.2 cm cyst in the right posterior lobe of the liver, a 1.2 cm × 0.8 cm cyst in the right ovary, and two cysts in the spleen, the sizes of which are 7.4 cm × 4 cm and 4 cm × 2 cm, respectively, and were all excised during the surgery. This case suggests that SHC can be complicated by cysts in other organs resulting in multiple origin cysts or a subtype of the multicystic diseases.
5.2.2.2 Clinical Features
More than 200 cases of SHC have been diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, with the male to female ratio of about 1:2 and age range of 5–87 years old with an average age of 53.5, and the male to female ratio is 1:4. Patients with small lesions may have no obvious clinical symptoms and were identified in physical examination or during abdominal surgery. Larger lesions may lead to slight upper abdominal discomfort, abdominal distention, hepatomegaly, and compression-related symptom. And complications of SHC are very common, including inferior vena cava thrombus [52], Budd-Chiari syndrome [53], fever and elevated white blood cells in intracystic infection, acute abdomen due to cystic rupture and reversion, jaundice in cyst-induced obstruction of the bile duct, and so forth. CT images show round hypodensity cysts with clear and smooth boundaries and should be differentiated from the image of parasitic diseases when both of them show calcification in the cystic walls [54].
5.2.2.3 Gross Features
Typical SHC are solitary monolocular cysts, several millimeters larger than 20 cm in diameter. In a few cases, the SHC are 40 cm in diameter, or multiple cysts, or even solitary polylocular cysts. The cystic walls are thin and smooth (Fig. 5.20). The content in the cysts is several milliliters to more than 10 liters in volume, and the fluid is often clear and canary yellow or mucous, while in certain cases the content is coffee or purulent due to hemorrhage or infection. If the cysts communicate with micro-bile ducts, the intracystic fluid may contain bile.
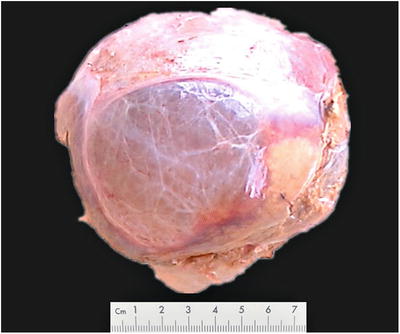
Fig. 5.20
Simple hepatic cysts, the cyst wall was thin and bright
5.2.2.4 Microscopic Features
The inner layer of the cystic wall is lined with simple low cuboidal epithelium, similar to the epithelium of bile ducts, with or without mucin, and the outer layer of the cystic wall is dense fibrous tissue, containing proliferated mucous glands and capillaries (Fig. 5.21), with intrahepatic biliary hamartoma in some cases.
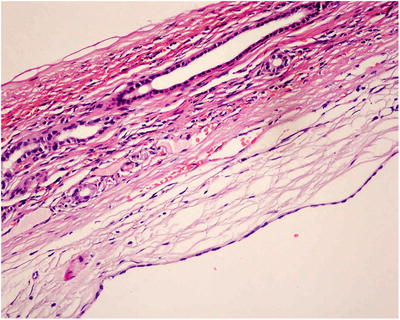
Fig. 5.21
The inner layer of the capsule wall was covered with a single layer of short cubic epithelium, and the lower layer was fibrous tissue
5.2.2.5 Immunohistochemistry
Epithelium is positive in the immunohistochemistry examination of CEA and EMA.
5.2.2.6 Differential Diagnosis
SHC are generally easily differentiated from tumorous lesions. However, it is difficult to differentiate SHC from malignant cystic tumors or liver abscess when hemorrhagic or purulent fluid is obtained via puncture. In extremely rare cases, canceration of SHC is observed, such as squamous carcinoma, adenocarcinoma (cystadenocarcinoma ), and mixed adenosquamous carcinoma (mucoepidermoid carcinoma), and much attention should be paid to sampling and microscopic observation.
5.2.2.7 Treatment and Prognosis
Fukunaga et al. [55] reported one case of a 25-year-old male patient with typical images of SHC without treatment [55]. But the cyst gradually enlarged and was complicated by calcification during the next 11 years’ follow-up, and the cystic wall was obviously thickened with papillary nodules on the inner wall. The cyst was excised via surgery and was diagnosed as biliary cystadenoma . Therefore, large cysts may be complicated by reversion, bleeding, rupture, compression on the bile duct, hyperplasia, or canceration and need surgical treatments [56–58].
5.2.3 Polycystic Liver Disease
5.2.3.1 Pathogenesis and Mechanism
Polycystic liver disease (PLD) demonstrates multiple cysts with varying sizes distributed in the whole liver, and the cystic wall was lined with biliary epithelium. In perspective of histogenesis, PLD belongs to the category of noncommunicating cysts, without communication with biliary tree. And the genesis of PLD involves malformation of embryonic bile duct plates of intrahepatic biliary tree, persistent segmentation, and cystic dilatation of the residual embryonic bile duct plates. Meanwhile the disconnection of the plates and distal bile ducts leads to retention of secretion and increased pressure in the ducts, and the cysts are gradually enlarged ending with PLD.
PLD is also known as autosomal dominant polycystic liver disease (ADPKD), classified into two types. One is polycystic kidney-related ADPKD in which ADPKD is complicated by autosomal dominant polycystic kidney disease (ADPKD). The other one is isolated polycystic liver disease (PCLD) without ADPKD. Among the 28 cases of PLD receiving surgery in Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, 24 patients (85.7%) were complicated by polycystic kidney disease with familial cystic diseases in 32% of them.
There are four identified PLD-related genes, among which PKD1 and PKD2 gene mutation exist in almost all ADPKD patients [59]. PKD1 gene is located on chromosome 16 (16p13.3), encoding polycystin PC1, and PKD2 gene is located on 4q22.1, encoding polycystin PC2. A great many reports on genetic mutation of ADPKD patients are found at home and abroad [60, 61], and no high frequency mutations of PKD1 and PKD2 genes have been discovered among their many mutations. Furthermore, about 20% of PLD patients have mutations of PRKCSH and SEC63 genes [62], encoding hepatocystin and SEC63 proteins [63], respectively. Since a proportion of PLD patients are without the mutations of PRKCSH and SEC63 genes, it is necessary to investigate the relation between other genes and PLD.
5.2.3.2 Clinical Features
The progression of PLD is slow and occult. No significant symptoms or signs in the early stage of PLD until the patients grow to middle or elderly ages, and the onset age of PLD is advanced from generation of generation. Among the 28 cases of PLD diagnosed in the Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, the male to female ratio is 1:4.6 with an average age of 51.9 years old. And it has been noticed that the numbers and volumes in female patients are larger, and PLD in women with multiple births or replacement treatment of estrogen are severer, suggesting the relation between estrogen and PLD, and estrogen may be one of the factors facilitating the development of PLD [64, 65]. Anjia Yang et al. (1995) reported a case of familial polycystic kidney and polycystic liver diseases, with ten patients among 12 people in three generations, and six patients had both polycystic liver disease and polycystic kidney disease. The age of onset was around 45 years in the first generation, 20–48 years in the second generation, and 5–13 years in the third generations. Clinical manifestations are related to the size of the cysts, and abdominal distention, abdominal pain, and hepatomegaly are the most common symptoms. And the compression of cysts on the adjacent organs or tissues results in the failure of these organs and other related symptoms. For example, compression of cysts on the hepatic duct or common bile duct causes jaundice, compression on the gastrointestinal ducts causes decreased appetite, and compression on inferior vena cava and hepatic vein results in ascites and lower extremity edema. In these cases, differential diagnosis should be made between PLD and malignant tumors. The clinical manifestations in the late phase of PLD as well as the complications are predominantly induced by hepatomegaly.
5.2.3.3 Gross Features
The lesions involve the whole or certain lobes of the liver, presenting as vesicular cysts in varying sizes (Fig. 5.22). On the section, the cyst wall is very thin and multilocular cavity with dark brown or clear fluid content can be seen.
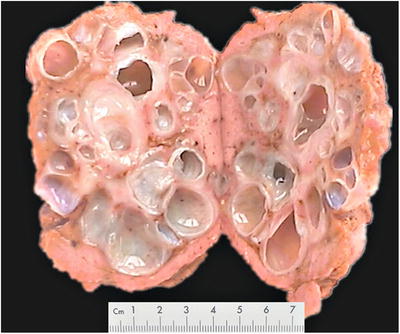
Fig. 5.22
Polycystic liver disease, the liver section with cysts of varying sizes, clear liquid inside
5.2.3.4 Microscopic Features
The inner layer of the cystic wall is covered by simple squamous epithelium or simple cuboidal epithelium, with positive CK staining, while the outer layer is fibrous tissue. The intercystic space is occupied by liver tissue, and no bile is observed in the cystic cavity due to its disconnection with bile ducts. And biliary hamartoma can be observed in the surrounding liver tissue (Fig. 5.23).
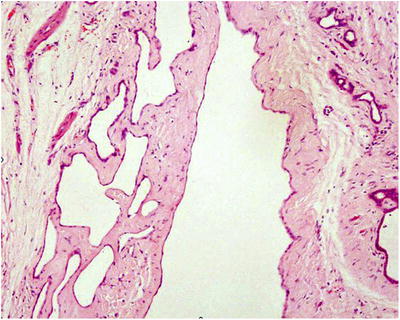
Fig. 5.23
Multiple cavities of varying sizes in the lesion, covered with a single layer of flat or cuboidal epithelium, and hyperplastic small bile duct could be seen in the cystic wall
5.2.3.5 Differential Diagnosis
PLD should be differentiated from multiple simple hepatic cyst and lymphangioma in the liver parenchyma. The latter is rare but similar to the lesions of PLD, as they are both multiple cysts, several millimeters to several centimeters in diameter, with or without other organ involvement. The key to identify a lymphangioma is the lymph in the cystic cavity with simple endothelium on the cystic wall which is D2-40 positive and no complication of multicystic kidney.
5.2.3.6 Treatment and Prognosis
Surgical treatments include cystic drainage and sclerotherapy, laparoscopic cyst fenestration, liver resection, and liver transplantation [66].
5.2.4 Caroli Disease
5.2.4.1 Pathogenesis and Mechanism
Caroli disease (CD) was first reported by a French researcher Jaequc Caroli in 1958, also known as congenital segmental cystic dilatation of intrahepatic bile duct, characterized by multicystic dilatations in the large intrahepatic biliary tree, especially the left and right hepatic ducts, segmental bile ducts, and branches. The dilatated bile ducts are communicated with branches of the intrahepatic bile ducts and belong to communicated cavernous dilatation of bile ducts. CD is now considered as an autosomal recessive genetic disease, and Caroli syndrome (CS) refers to the combination of CD and congenital hepatic fibrosis. The incidence of CD is 1/6000–1/400000 in newborns, who may have other congenital disorders. And an autosomal recessive genetic disease has an incidence rate of 1/20000 in newborns, closely related to Caroli syndrome. A case of a 2-month-old male infant has been reported to have Caroli disease accompanied by a rare disease called vein of Galen malformation [67]. Furthermore, mutation of PKHD1 gene was discovered in the whole exome sequencing in familial cases of Caroli disease [68].
5.2.4.2 Clinical Features
CD mainly occurs in children or adolescent, and the proportion of patients with the onset age under 10 years old is about 60%. The predilection age range is 21–49 years, and 75% of the patients are male. Caroli disease is classified into two subtypes. Type I is simple CD, with multicystic dilatations of intrahepatic bile ducts, often combined with intrahepatic stones of bile ducts, and has clinical manifestations such fever, abdominal pain, and recurrent biliary infection. Type II is CD with diffuse or periportal fibrosis and heperplasia of fibrous tissue surrounding the portal area with proliferated small bile ducts, and early occurred hepatosplenomegaly, portal hypertension, and esophageal variceal rupture and bleeding are also observed. The clinical manifestations of CD are often nontypical, including symptoms and signs caused by inflammation and stones due to dilatation and bile retention of the intrahepatic bile ducts, with a wide range of onset age. Patients of simple CD may have decreased appetite and body weight, recurrent right abdominal pain, and fever, with or without jaundice, and patients accompanied by cholangitis present deepened jaundice. In a few cases, recurrent jaundice is the main symptom. And in patients with periportal fibrosis, clinical manifestations are mainly portal hypertension, splenomegaly, and upper gastrointestinal hemorrhage.
5.2.4.3 Pathological Features
The main histological features include: (1) hyperplasia of small portal bile ducts, with dilated ductular lumen and hyperplasia of fibrous tissue in the wall, (2) branches of portal vein in the portal area with fibrous bridges between portal areas, and (3) normal hepatic lobules. Chronic inflammation of biliary mucosa, caused by bile retention and biliary stones in the cystic dilatated bile ducts, often results in biliary epithelial hyperplasia, metaplasia, or dysplasia, and the cystic wall appears thick and rough, thus forming cystic lesions (Fig. 5.24). Under microscope, obvious hyperplasia of the inflammatory fibrous tissue can be observed, with involvement of hyperplasia of fibrous tissue and small bile ducts surrounding the portal area, similar to the changes in sclerosing cholangitis and early phase of liver cirrhosis. Canceration occurs in 7–14% of CD cases, 100 times the risk of healthy people, which is also observed in our pathological work (Fig. 5.25), thus is considered as a precancerous lesion of cholangiocarcinoma .
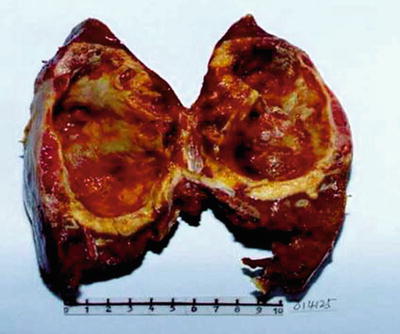
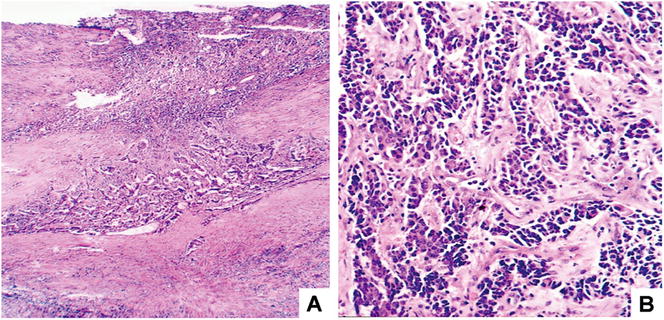
Fig. 5.24
Caroli disease bile duct dilatated into a large cystic cavity, inner wall rough, and thickening
Fig. 5.25
Canceration of Caroli disease. (a) Small piece of cancer in the center of the cyst wall; (b) poor differentiation of cancer cells, beam and cable-like arrangement, invasive growth
5.2.4.4 Treatment and Prognosis
Type I CD can be treated with surgical resection, and type II CD can be treated by liver transplantation.
5.2.5 Congenital Choledochal Cyst
5.2.5.1 Pathogenesis and Mechanism
Congenital choledochal cyst (CCC) is characterized by cyst or fusiform dilatation of the common bile duct, with or without dilatation of intrahepatic bile ducts, also known as dilatation of the common bile duct, which is the most common congenital abnormality and most common disease of congenital hepatobiliary cysts, with an incidence rate of 1/1000–1/190000 and a male to female ratio of 1:3–1:4. It is generally believed that the incidence of CCC in the Asian population is significantly higher than that in European and American populations.
5.2.5.2 Clinicopathologic Features
CCC is commonly discovered in infants and children under the age of 10, but its age range covers any ages. More than 200 cases of CCC have been diagnosed in our Department of Pathology, Eastern Hepatobiliary Surgery Hospital, Second Military Medical University, in the last 5 years, with a male to female ratio of 1:3, and the average age of the patients was 51 years old ranged 3–76 years. The clinical manifestations include abdominal pain, jaundice, and upper abdominal mass, which are known as a triad. Pathologically, dilatation of the common bile duct or extrahepatic bile ducts is observed, sometimes forming a diverticulum (Fig. 5.26). Large cysts may perforate or rupture spontaneously [69, 70]. Intrahepatic bile duct cyst containing thick bile can also be observed with a thickened wall consisting of dense collagen fibers, scattered smooth muscular fibers, abnormal glands, and inflammatory cells. And epithelium can rarely be seen in the wall of intrahepatic bile duct cyst.
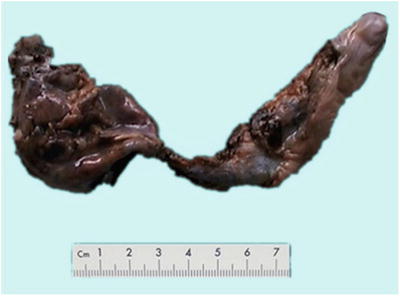
Fig. 5.26
Congenital choledochal cyst, cystic dilatation of the common bile duct, the diameter of bile duct was significantly increased
The classical and widely used classification is proposed by Todani and divided CCs into five types:
Type Ia
cystic dilatation of the common bile duct
Type Ib
segmental dilatation of the common bile duct
Type Ic
fusiform dilatation of common bile duct
Type II
diverticulum of common bile duct formed by outward convex of the bile duct wall
Type III
prolapsed cyst at the end of the common bile duct
Type IVa
combination of the cyst of the common bile duct and dilatation of intrahepatic bile duct
Type IVb
multiple dilatation of extrahepatic bile ducts
Type V
single dilatation of intrahepatic bile duct or multiple cysts (Caroli disease).
In Eastern countries, type IV and type V are the most common CCs. The risk of canceration of CCs is 2.5–15%, and secondary chronic inflammation increases the risk of canceration of CCs by more than 20 times.
5.2.5.3 Treatment and Prognosis
Due to the secondary repeated infections, biliary cirrhosis, bile duct perforation, or canceration complicated to CCs, timely surgical treatments are recommended once the diagnosis of CCs is made. The principle of surgery is to reconstruct the bile drainage into the intestine to prevent retrograde cholangitis and excise the dilatated common bile duct to prevent canceration.
5.2.6 Congenital Hepatic Fibrosis
5.2.6.1 Pathogenesis and Mechanism
Congenital hepatic fibrosis (CHF) was named by Kerr in 1961 and is an important type of hepatic fibrous cystic diseases. The mechanism involves the developmental malformation of biliary plates, and damage occurs mainly in the interlobular bile ducts. CHF is an autosomal recessive hereditary disease, and a domestic report in China concerned a pair of siblings, brother and sister, who both had CHF. CHF is commonly complicated by congenital intrahepatic biliary dilatation (Caroli disease) and polycystic kidney disease, and other complicated diseases include medullary sponge kidney, Ivemark familial dysplasia, Meckel syndrome, vaginal atresia, and less common adult diseases such as polycystic kidney disease and tuberous sclerosis [71, 72]. Al Sarkhy et al. [73] reported a case of 4-year-old boy with Prader-Willi syndrome and CHF [73], suggesting that CHF may be related to the rare hereditary disease induced by abnormal chromosome 15. In addition, Kinugasa et al. [74] reported a case of CHF complicated with HCC [74], while Paradis et al. [75] reported a case of CHF with multiple hepatocellular adenomas [75].
5.2.6.2 Clinical Features
CHF mainly occurs in perinatal, newborn, infant, and juvenile phases, and most patients are aged 1.8–14 years old with an average of 15 years. Patients with symptoms are most 3–6 months old, and males are slightly more than females. The clinical manifestation primarily includes liver cirrhosis and portal hypertension (70%), and hepatosplenomegaly can be found in infant patients, and later hematemesis, hematochezia, esophageal varices, etc. The major clinical feature for CHF is normal hepatic function in the patients. CHF can be divided into four types: portal hypertension type, cholangitis type, mixed type, and occult type.
5.2.6.3 Gross Features
CHF clinically manifests enlarged, hard liver, without visible vesicles or with microvesicles formed by dilatated bile ducts; thus, it is also known as microcystic hepatic disease, communicated with intrahepatic biliary tree, and is a type of communicating cysts.
5.2.6.4 Microscopic Features
Histological change of CHF is the damage on the interlobular bile ducts, including:
- 1.
A large amount of fibrous tissue hyperplasia surrounding the portal area, forming a broad fibrous septa inter portal areas, the portal areas expanded, with no or mild inflammation.
- 2.
Large amounts of different forms of proliferated small bile ducts in the fibrous septa.
- 3.
The septa or fibrous bundles stretch or segment the lobules, with no pseudolobuli, and the structure of lobules maintains normal (Fig. 5.27).
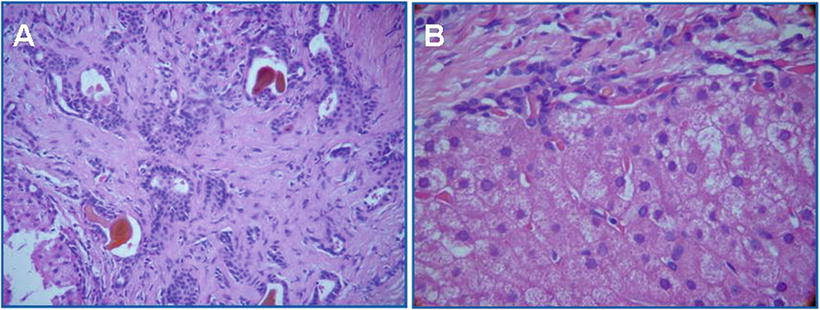
Fig. 5.27
Congenital hepatic fibrosis. (a) Interstitial substance small bile duct hyperplasia with cholestasis; (b) liver cells have no obvious inflammation or necrosis, portal area fibrosis (Cite from Abdul Wahab A [76])
- 4.
The central veins remains in the center of the hepatic lobules, suggesting the microcirculation of hepatic lobules remains normal.
5.2.6.5 Treatment and Prognosis
The prognosis is related to the severity of portal hypertension in CHF patients, and the fatality rate induced by complications of CHF reaches 50%, among which sepsis, gastrointestinal hemorrhage, or severe nephropathy are important prognostic factors. Due to the commonly seen cholangitis, there is risk of intrahepatic cholangiocarcinoma in the patients of CHF, who often need shunting or devascularization due to portal hypertension, with relatively good prognosis if portal hypertension is well controlled. For patients with severe liver fibrosis and portal hypertension, liver transplantation is the best therapy.
Supplementary: Hepatobiliary Fibrocystic Disease
Hepatobiliary fibrocystic disease (HFD) refers to a group of congenital hereditary diseases, characterized by biliary dilatation and liver fibrosis of intra- and extrahepatic bile ducts and portal areas. Aforementioned biliary hamartoma (microbile duct), polycystic liver disease (microbile duct), congenital hepatic fibrosis (microbile duct), Caroli disease (micro, medium, or large bile duct), and congenital biliary duct cyst (extrahepatic large bile duct) are all members of HFD, sharing the common pathogenesis of malforma.
The development of biliary system includes intrahepatic and extrahepatic parts, both of which converge into interlobular bile ducts in the portal areas. In the early phase of fetal development, biliary plates are bilayered cylindrical structures. In ordinary conditions, in the ninth week of fetal development, hepatoblasts are located around intrahepatic portal veins and branches like sleevelet, forming the primary biliary plates which are the blastema of the development of intrahepatic biliary system. At the twelfth week in fetal development, the reconstruction of biliary plates begins, the primary biliary plates are replaced by ducts circled by interstitial cells, and the epithelium of bile ducts is replaced by cuboidal or columnar epithelium, stretching into the hepatic lobules forming intralobular bile ducts, and further construct the portal ductular system with portal vein and hepatic arteries. By 1 month after birth, the reconstruction of biliary plates has finished, and the residual biliary plates degenerate and disappear. Otherwise, the development of biliary system stagnates, and more embryonic biliary plates retain to form malformation of biliary plates, manifesting large amounts of ductular or cystic dilatation of proliferated small bile ducts in the portal areas with irregular lumen and surrounding fibrous tissue. In malformation of small bile ducts, CD10, CD7, and MUC-1 staining are positive. On the other hand, congenital cysts of the common bile duct manifest malformation of extrahepatic major bile ducts. As to the proliferated small bile ducts in FHD, it should be noted that liver masses predominant in HFD be differentiated from tumors according to clinical, imaging, pathological evidences [77].
5.2.7 Ciliated Hepatic Foregut Cyst
5.2.7.1 Pathogenesis and Mechanism
Ciliated hepatic foregut cyst (CHFC) is a congenital hepatic cyst originating from the embryonic foregut, commonly seen in the spleen. It was first reported by Friedreich (1857) as ciliated hapatic cyst and later was given the name of CHFC by Wheeler and Edmondson. In the perspective of histogenesis, CHFC was developed from residual embryonic foregut and differentiate into bronchial tissue in the liver, forming CHFC. The epithelium of CHFC is similar to bronchial mucosa, and Clara cells with positive CD10 have also been observed and reported. By 2014, more than 100 cases of CHFC have been reported in English literature, and around 100 cases of CHFC have been reported in Chinese literature [78].
5.2.7.2 Clinical Features
The patients of CHFC have a male to female ratio of 1.1:1, with an average age of 50 years old aged from 3 months to 82 years. Most of them are discovered accidentally and sometimes manifest upper abdominal pain, obstructive jaundice, and portal hypertension. The level of serum CA19-9 elevates in patients with large cysts. And the cysts in children manifest large masses, due to its communication with bile ducts leading to retention of bile in the cysts. Ultrasound B examination displays hypoechoic lesions with clear boundaries. CT images demonstrate hypodensity with no enhancement after administration of contrast reagent. And CHFC exhibits hyperintensity on T2-weighted images and hyperintensity in most cases on T1-weighted images in MRI. In one third of the cases, the radiation intensity of the lesions varies according to the different mucin concentrations and calcium levels in the cysts [79].
5.2.7.3 Gross Features
The lesions of CHFC are often located in the middle of the left lobe of the liver superficially, restricted under the liver capsule. A patient may have single or multiple cysts, and monolocular cysts are more commonly seen with mucous content. The lesions are usually small with an average volume of 3.6 cm in diameter, while some lesions are larger than 10 cm in a few cases. Monolocular cystic masses are predominant, often 1–4 cm in diameter with an average of 3.6 cm, and the content of the cysts is bile-like fluid, with thin and smooth inner layered walls, and valve-like strips can be observed regionally (Fig. 5.28). And in a domestic case of CHFC, the 8-year-old boy had a 30 cm in diameter lesion and was treated via fenestration and drainage for the cyst.
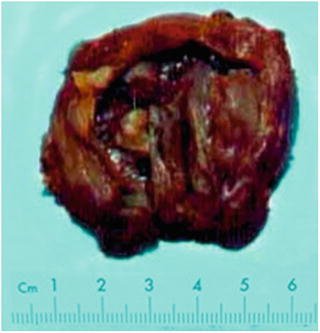
Fig. 5.28
Ciliated hepatic foregut cyst, multilocular cyst with smooth inner wall
5.2.7.4 Microscopic Features
The cystic wall of CHFC can be divided into four layers from top to bottom: (1) pseudostratified ciliated columnar epithelium with slender and evenly arranged cilia on the top of the cells and a few amount of goblet cells in the epithelium (Fig. 5.29), and the cilia is positive in actin, CK, CEA, and EMA staining; (2) lamina propria, which is a loose connective tissue beneath the epithelium; (3) smooth muscular layer; and (4) the outer layer of fibrous tissue, which contain thick-walled vessels and proliferated small bile ducts. The content of the cysts includes neutral or acidic mucus and cellular debris.
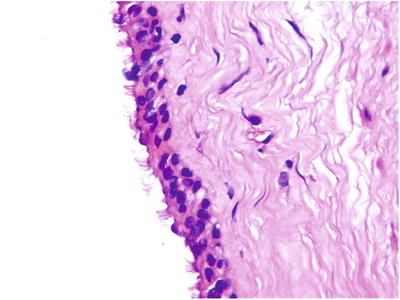
Fig. 5.29




The cyst wall was covered with pseudostratified ciliated columnar epithelium with cilia that appears on the apical
Related posts:

Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
