Groups of tumor antigens
Antigens
Cancer type
Cancer testis (CT)
MAGE1-3 and -6
Melanoma, breast, head/neck
HAGE, GAGE, NY – ESO – 1, BAGE, XAGE
Bladder, gastric and lung, head/neck, many cancers
Differentiation antigens
Tyrosinase, gp- 100, TRP-1, and -2, MART-1
Melanoma
NY-BR1
Breast cancer
Viral antigens
EBV
Burkitt’s lymphoma
HepB
Hepatitis B
HPV
Cervical and penile cancer
HTLV
T-cell leukemia
Oncofetal antigens
CEA
Colon, breast, pancreatic
α-fetoprotein
Liver cancer
5T4
Many carcinomas
Oncotrophoblast glycoprotein
Many carcinomas
Tumor-specific antigens
CDK4
Melanoma
Caspase-8
Head/neck
b-catenin
Melanoma
BCR/ABL
CML
p53 (mutated)
Breast, colon, other cancers
Ras (mutated)
CML, AML, ALL
Overexpressed/mutated antigens
HER-2/neu
Breast, ovary, lung
MUC-1
Breast, adenocarcinoma colorectal
p53 (nonmutated)
Lung, bladder, head/neck
WT-1
Pancreatic, colon, lung
Proteinase-3
CML
PAP, PSA, PSMA, survivin
Prostate
Idiotype antigen
Ig idiotype
B-cell NHL, MM
Major histocompatibility molecules (MHCs) present on the cell surfaces allow the immune system to distinguish between self, modified, or nonself antigens. MHCs present cleaved protein fragments in the form of peptides following processing via the proteasome (class I) or the endosomal compartment (class II). Class I peptides induce cytotoxic effector T cells (CTLs) with specificity against tumor antigens, whereas class II peptides induce T helper (Th)-mediated immune response, which are also important in assisting the development of CTL memory. Hence it is recognized that both class I and class II epitopes should be included in peptide vaccine strategies.
Following the first report in 1991 that vaccinations with a single MHC class I binding CTL peptide epitope in IFA-protected mice against a subsequent challenge, many studies focused on the efficiency of this mode of vaccination [2]. This method proved to be beneficial in some preclinical models, for example, protection against the outgrowth of HPV16 in mice, but failed to show a good clinical correlation. Further studies on the peptide vaccination strategy showed that increasing the length of the peptide to include multiple CTL and Th epitopes significantly enhanced the efficiency of peptide vaccinations. The use of peptides where the anchor residues are substituted or “mimotopes” to enhance MHC–antigen interaction is a current strategy [3].
It has been observed that bulky tumors (developed at later stages of cancer) elicit tolerizing conditions within their tumor microenvironment, providing an escape mechanism for the tumor. Tumors are also responsible for suppression of immunosurveillance which inhibits the local antitumor immune response. Suppression of the immune system takes place through different mechanisms such as impairment of antigen presentation, activation of negative co-stimulatory signals, active biosynthesis of immunosuppressive molecules, recruitment of regulatory T cells (Tregs), and transformation of T cells locally into Tregs. Thus, suppressor cells produced within the tumor can migrate to lymph nodes and can give rise to immunosuppression which may represent an important mechanism for failure of immunotherapies in clinical trials. Using adjuvants/agonists has provided a new avenue, leading to the improvement in recurrence-free survival. This allows us to conclude that vaccination at early stages of disease progression is advantageous for inducing a stronger antitumor immune response.
Improved methodologies have significantly contributed to tumor antigen identification and assessment of functionality. The most commonly used techniques can be broadly divided into two, namely, the reverse immunology approach and the direct approach. In this review we consider these approaches in some detail, outlining the basis for their use. Finally, we will discuss the way in which immunogenic peptides derived from tumor antigens are being used in cancer immunotherapy trials.
4.2 Reverse Immunology Approach to Peptide Identification
The steps involved in this approach are well established (Fig. 4.1) and are less time consuming compared to the direct approach (Fig. 4.2). Firstly, candidate genes are identified/selected based on tumor-restricted expression. This can be achieved using PCR assays to determine RNA expression levels or antibody staining of cancer vs. normal tissues to identify differentially expressed antigens. Secondly, immunogenic peptide epitopes of cancer antigens are predicted by in silico analysis using several different computer-based algorithms. On successful validation on the immunogenicity of the identified peptide epitopes in vitro and in vivo, they would be selected for clinical trials. To date, reverse immunology has resulted in the identification of several MHC class I and class II peptides, which are recognized by antigen-specific T lymphocytes. These include peptides derived from MAGE-1, MAGE-2, MAGE-3, TRP2, gp100, HER-2/neu, SSX-2, PRAME, and EphA.
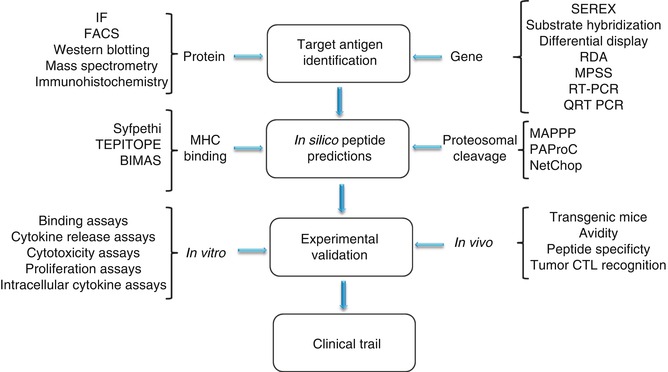
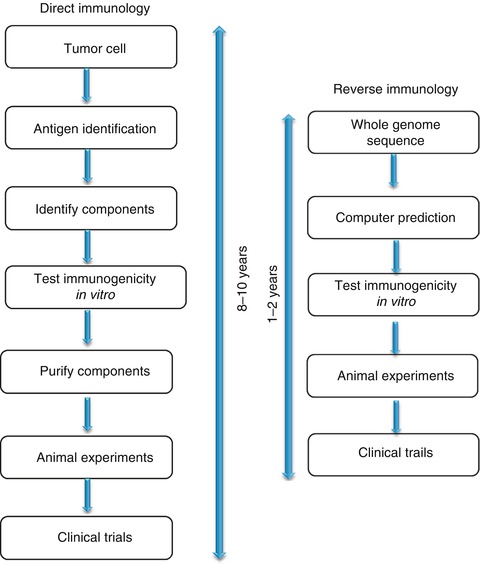
Fig. 4.1
Steps involved in reverse immunology approach
Fig. 4.2
Comparison of direct and reverse immunology approaches. Reverse immunology is well established and less time-consuming compared to direct approach
4.2.1 Target Antigen Identification
The identification of cancer antigens for immunotherapy represents a crucial step towards clinical immunotherapy. Tumor antigens are divided into tumor-unique antigens, whose expression is restricted to tumor tissue- or tumor-associated antigens which are usually overexpressed in tumors, but may show low levels of expression in normal tissues. In functional terms, TAs can be broadly divided into antigens that are required for tumor development and progression (indispensable antigens) and those antigens that are nonessential. Immunotherapy approaches targeting antigens that are not crucial for tumor development may eventually result in antigen-loss variants arising within the tumor, eventually leading to tumor escape. Therefore an ideal TA candidate would be quintessential for tumor development and would also be expressed in a wide variety of tumors making it a “Universal TA” [4]. The National Cancer Institute recently conducted a program to prioritize cancer antigens to establish a list of “well-vetted,” priority ranked TA targets based on predefined unprejudiced criteria [5]. Adopting a pairwise approach, the criteria weighting for antigens, in descending order, was as follows: (a) therapeutic function, (b) immunogenicity, (c) role of the antigen in oncogenicity, (d) specificity, (e) expression level and the percentage of antigen-positive cells, (f) stem cell expression, (g) number of patients with antigen-positive cancers, (h) number of antigenic epitopes, and (i) cellular location of antigen expression [5].
The initial step in reverse immunology is the search for protein/gene expression patterns selectively observed in tumor cells. Protein overexpression could be detected by using techniques such as immunofluorescence, western blotting, flow cytometry, etc. The shortcoming of these techniques is that they are time consuming and depend on the availability of antibodies with high sensitivity and specificity. Also, with this method it is difficult to estimate protein turnover [6]. New large-scale gene expression assays such as cDNA microarrays, oligonucleotide chips, cDNA library sequencing, serial analysis of gene expression (SAGE), massively parallel signature sequencing (MPSS), subtractive hybridization, differential display PCR, and representational difference analysis (RDA) are a few of the techniques widely used to decipher complex expression patterns and identify new candidate antigens. SEREX (serological analysis of recombinantly expressed clones) has been a widely used technique that relies on the use of cancer patients’ sera to screen selected tumor cDNA libraries [7, 8]. Tumor antigens identified via this method usually contain CD4+ T-helper lymphocyte epitopes. The first tumor antigen, MAGE-1, was discovered through autologous typing and application of a newly developed DNA-cloning technique for defining the targets of T-cell recognition [9]. A melanoma patient with unusually favorable clinical course was identified to have CTLs that recognized autologous tumor cells. With antigen-specific T cells as a reagent and through the use of cosmid gene libraries, it was possible to identify and clone the MAGE-1 gene. Studies on MAGE-1 showed for the first time that the human immune system can respond to TA, and the findings transformed the study of tumor antigens and stirred a dynamic effort to discover tumor antigens, which has resulted in a long and still-growing list of antigens from a variety of tumors which potentially serve as targets for immunotherapy.
The use of antigenic epitopes/peptides to promote antitumor immunity represents one of the simplest and most applicable ways of targeting cancer cells expressing the respective protein. Recent studies have shown that immunotherapy approaches targeting multiple epitopes at the same time [10] or use of long synthetic peptides that would comprise multiple epitopes [11] can result in delivering clinical benefits to a greater number of patients. This strategy would broaden the clinical response in several ways:
1.
In principle this would stimulate both CTL and T helper cell epitopes simultaneously.
2.
Targeting different HLA types will increase the number of patients eligible for the vaccine.
3.
This approach would decrease the risk of immune escape by tumor cells.
4.
In addition, it would allow the synthesis of an “off-the-shelf vaccine” that could be used for different types of tumors.
4.2.2 In Silico Peptide Predictions
A number of computer-aided tools have been developed for the prediction of T-cell epitopes. These algorithms are based on the natural processing and presentation of proteins; the number of computer-aided algorithms available for the prediction of better T-cell epitopes reflects the existing level of understanding antigen processing. Thus, the tools designed for T helper epitope prediction are comparatively less advanced than for CTL epitope prediction.
For a target protein to be successfully presented on an MHC class I or class II molecule, it must undergo a number of processing steps resulting in the transport and cleavage of the peptide. Epitopes presented on MHC class I molecules are 8–11 amino acid length chains and are predominantly derived from intracellular proteins. A cytosolic multi-subunit proteolytic complex, known as the proteasome, degrades proteins to peptides which are later transported into the endoplasmic reticulum (ER) by the adenosine triphosphate (ATP)-dependent transporters associated with antigen processing (TAPs) [12]. The alpha chain of MHC I binds to the beta-2 microglobulin unit with the help of calnexin. Once stable, calnexin is replaced with calreticulin and tapasin [13]. Within the ER, peptides undergo further N-terminal trimming before their subsequent loading into the empty MHC-binding cleft. The epitope binds tightly to the epitope-binding cleft stabilizing the trimeric complex transported to the cell surface via the ER and Golgi network [13]. Exogenous proteins are processed mainly by the MHC class II pathway. The alpha and beta subunits of the MHC class II molecule are preoccupied by a non-polymorphic invariant chain (Ii) which acts as a chaperone for class II folding and prohibits binding of intracellular proteins to MHC II [14]. The extracellular proteins are engulfed by endosomes which transport them to the Golgi apparatus. The inactive MHC II molecule is also transported to the Golgi where proteolytic degradation of Ii occurs, leaving the class II-associated invariant peptide (CLIP) in the peptide-binding cleft. The MHC II–CLIP complex can then interact with human leucocyte antigen (HLA)-DM (H-2M in mouse) which activates the dissociation of CLIP, allowing the loading of peptides into the empty MHC class II cleft [14]. The development of software prediction algorithms is based on peptide–MHC interactions or on proteasomal degradation.
4.2.2.1 Peptide–MHC Interactions
This is one of the early prediction tools developed and is based on the fact that MHC molecules would bind to peptides with similar “motifs.” This fundamental principle led to the development of computer-based algorithms which screen potential peptide sequences of defined lengths and with similar binding motifs.
Broadly, the MHC-binding peptide prediction methods can be divided into three main groups: (a) motif-based methods, (b) statistical/mathematical expression-based methods, and (c) structure-based methods. Motif-based methods consider every amino acid within a peptide and assign it a positive or negative value, depending on the characteristics of the MHC groove with which it will interact [15]. SYFPEITHI (www.syfpeithi.de) is one of the widely used evidence-based motif matrix, as the data used within the algorithm are derived from the knowledge of actual natural ligands and can predict both class I and class II epitopes [16]. Another matrix-based prediction tool widely used is TEPITOPE (www.vaccinome.com), in which matrices are constructed based on the interaction of every amino acid with the MHC-binding cleft [17]. Nonetheless, instead of determining this empirically for each HLA allele, it combines these data with HLA sequence variation data to form virtual matrices. Even though the program is restricted to MHC class II, it allows prediction of highly promiscuous peptides within one search [17]. Structure-based methods calculate the binding energy of the peptide–MHC complex, and peptides energetically favored are predicted as binders. BIMAS is such a prediction system (http://bimas.cit.nih.gov/) that generates results expressed as estimated peptide dissociation values [18]. Structure-based methods utilize the power of artificial neural networks. The predictive accuracy of this method is very high, but these are more complex, nonlinear self-learning systems and require large amounts of data for learning [18]. PREDICT (http://sdmc.lit.org.sg:8080/predict/) and nHLAPred (http://www.imtech.res.in/raghava/nhlapred/) are examples of epitope selection methods.
4.2.2.2 Proteasomal Degradation
As described earlier, the proteasome is charged with recycling proteins and, hence, plays a major role in deciding whether a peptide is likely to be available to bind to MHC molecules. The proteasome has at least three different catalytic activities: trypsin-like (cleavage after basic amino acids), chymotrypsin-like (cleavage after hydrophobic amino acids), and peptidyl-glutamyl peptide-hydrolyzing activity (cleavage after acidic amino acids) [19]. The overall enzymatic activity (cleavage, inhibiting or enhancing) is a result of interaction between all subunits making the process complex. At present, three proteasome cleavage prediction methods are publicly available: PAProC (www.paproc.de) developed by Tubingen University, MAPPP (www.mpiib-berlin.mpg.de/MAPPP/) developed at the Max Planck Institute in Berlin, and NetChop (www.cbs.dtu.dk/service/NetChop/) developed at the Center for Biological Sequence Analysis at the Technical University of Denmark. Prediction Algorithm for Proteasomal Cleavages (PAProC) is a method for predicting cleavages by human and yeast (wild-type and mutant) proteasomes [20]. The influence of different amino acids at different positions is assessed using a stochastic “hill-climbing” algorithm. The PAProC server also allows the identification of peptides cleaved by the immune proteasome [21]. This is highly advantageous since current data suggest that some tumor antigens, such as MAGE-3 (melanoma-associated antigen 3), would only be produced by the immune proteasome [22]. Also components of the immune proteasome have been found to be associated with tumor-infiltrating lymphocytes (TILs) in spontaneously regressing tumors. Though the complete role of the immunoproteasome has yet to be deciphered, selection of epitopes from both types of proteasomes will unveil the epitopes more suitable for immunotherapy [23].
MAPPP (MHC-I Antigenic Peptide Processing Prediction) is another approach that combines the proteasomal cleavage with MHC-binding prediction [24]. FragPredict is a component of the MAPPP package that deals with the proteasome cleavage prediction and consists of two algorithms; the first algorithm uses statistical analysis to predict potential cleavage sites, while the second uses results of the first algorithm as an input and predicts the fragments most likely to be generated. The second algorithm is based on the time-dependent degradation of a kinetic model of the 20S proteasome [24].
NetChop is a neural network-based method anchored on MHC class I ligands generated by the human proteasomes [25]. The rationale behind this approach is that every MHC ligand has to be generated by the proteasome; therefore, these ligands bear the closest resemblance to naturally processed in vivo cleavage products. The MHC class I ligands used to develop NetChop were compiled from public databases [20], two versions of which are available, 1.0 and 2.0, and the later version is trained with a data set that is three times larger.
4.2.3 Epitope Validation
In the validation phase of reverse immunology, the natural presentation and immunogenicity of the selected epitopes should be corroborated. Using cell lines or tumor tissues expressing the appropriate antigen and HLA allele, biochemical methods can be used to elute peptides from the cell surface or from isolated HLA antigen. The purified products are then analyzed by mass spectrometry [26] to derive sequence information and identify the target peptide (as discussed in detail below). Though this technique confirms the expression of HLA-bound ligands, it does not allow the assessment of peptides’ immunogenicity. Assessing the immunogenicity of predicted peptides relies on demonstrating their ability to stimulate MHC class I- or class II-restricted T-lymphocyte responses. These may be either a primary response, where naïve T cells respond to antigens in culture or secondary, where, for example, patient CD8+ or CD4+ T cells, already exposed to antigen in vivo, demonstrate a secondary response. However, patient response to self-(tumor) antigens is generally quite weak, and T lymphocytes may become tolerant towards this antigen. Tolerance may be overcome by exposure of patient lymphocytes to a combination of interleukin (IL)-2 and IL-12 in vitro, which enhance the tumor-specific CD8+ T-cell response and additionally prevent overgrowth of nonspecific, less-effective lymphokine-activated killer cells [27]. IL-12 is a potent inducer of tumor-specific CTLs and promotes the production of Th1 cytokines [28].
The immunospot assay, which is based on the detection of cytokine secretion in response to antigen, is used to detect antigen-reactive T-cell responses. Most current assays for measuring T-cell cytotoxicity are based on alterations in plasma membrane permeability and the subsequent release (leakage) of components into the supernatant (51Cr, lactate dehydrogenase assays) or the uptake of dyes (CFSE), which are normally excluded by viable cells. Another alternative is the use of flow cytometry to detect the expression of CD107 in the membrane, which is transiently expressed during the process of cell killing [29]. Use of cytokine-secretion assays, intracellular cytokine assays, HLA class I multimer staining (e.g., peptide-specific tetramers), etc. are among the techniques that have been thoroughly validated and established recently [30]. The use of tetramers has proved to be especially successful for the identification of peptide-specific CD8+ T lymphocytes and to a lesser extent for CD4+ T helper cells.
4.3 Direct Immunology Approach
The inherent weakness of reverse immunology is the incredibly low probability of identifying a peptide that is naturally processed, presented, and sufficient to induce CTL activity and tumor lysis. As a consequence, low-throughput attempts to screen limited numbers of peptides are typically unsuccessful. In addition, MHC peptides with low MHC receptor-binding affinities or those carrying post-translational modifications cannot be predicted with this approach. Hence, the laborious approach of direct biochemical isolation of T-cell epitopes still remains invaluable. Tumor cells (isolated from solid tumors or blood) or tumor cell lines could be used as a source for MHC–peptide isolation. Studies on tumor cell lines are advantageous due to their unlimited expansion capacity in vitro. However, the variations induced by in vitro passaging should be taken into consideration. Hence direct analysis of uncultured tumor cells should be performed where possible. The steps involved in direct immunology approach are (1) isolation and purification of peptide–MHC complexes, (2) analysis of purified epitopes, and (3) assessment of the immunogenicity of epitopes.
4.3.1 Isolation of Peptide–MHC Complexes
The techniques commonly used for isolation of MHC-associated peptides usually involve immunoaffinity chromatography and acid elution. Immuno-affinity chromatography (IAC) combines the use of LC with the specific binding of HLA antigens to antibodies or related agents [31]. The source material for this approach is usually frozen tissue, blood cells, or cultured cell lines. Solid tissue is first mechanically dissociated in the presence of protease inhibitors (to avoid any cleavage of MHC complexes) at a stable pH value (usually between pH 7.0 and 8.0). After washing (by centrifugation and filtration) the lysate is passed over MHC-specific monoclonal antibodies bound to sepharose beads. The beads are then washed to remove excess detergents, and MHC complexes are released from the antibodies by acid treatment. The peptides can be separated from the proteins by ultrafiltration, and the flowthrough is usually lyophilized before fractionation and sequence analysis. Though the technique provides highly pure isolates, it suffers disadvantages such as high cost due to the requirement of large amounts of antibodies (10–30 mg per isolation), complexity of the protocol, and inability to distinguish intracellular and extracellular MHC complexes [32]. Recent studies that tried to include desalting and inclusion of specific ions in desalting buffer have been shown to enhance peptide yield [33].
The acid elution technique is based on the release of MHC–peptide complexes from the cell surface by short acid treatment at pH 3.3 [34]. The source materials for this technique are cells from dissociated tissue or adherent or suspension cell cultures. Having intact cells is a prerequisite for the technique since cell damage will lead to the release of proteases generating peptide fragments from highly abundant cell proteins. The major advantage of the technique is that it is cost effective, simple, and it could differentiate intracellular and extracellular MHC complexes. This method has been successfully employed to identify T-cell epitopes from melanoma cells [35], as well as from the bcr–abl fusion protein expressed at the cell surface [36].
4.3.2 Analysis and Sequencing of MHC-Associated Peptides
HPLC fractionation can be performed prior to tandem mass spectrometry, which in combination allows high-resolution separation and sequencing of single peptides from complex samples [37]. Mass spectrometry (MS) is based on precise determination of molecular masses of analyte molecules. Following determination of the molecular mass of the analyte by various means (depending on MS instrumentation), the peptide sequence can be derived by fragmentation of the analyte ion. Hence MSMS analysis allows the detection of a single peptide from a complex mixture of peptide pools. Figure 4.3 illustrates an example of MSMS spectrum of peptides.
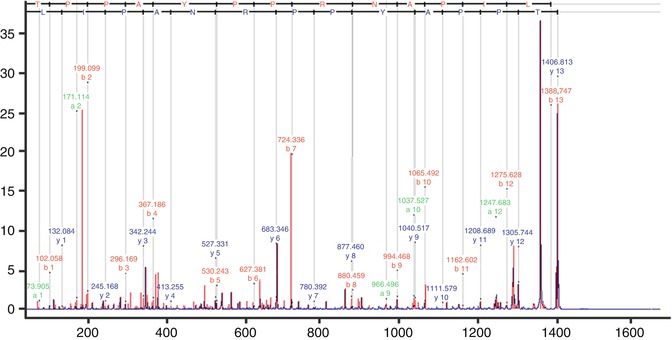
Fig. 4.3
MSMS spectrum showing HepB peptide of mass 1406.813. Fragment ions derived from the precursor (peptide ion) allow the assignment of the amino acid sequence of the peptide
4.4 Human Immunotherapy Against Tumor-Associated Peptides
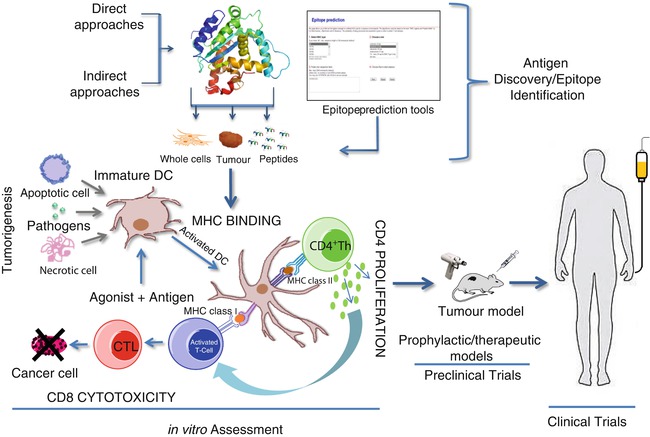
Identification of the tumor-specific antigens and peptide epitopes expressed on MHC class I antigens on the cancer cell surface has facilitated the development of new approaches to immunotherapy. These cancer-specific antigens possess the potential to be used in the vaccine-based therapies, targeting the respective antigen; however, extending life expectancy and survival are necessary criteria for FDA approval and acceptance of these treatments. Figure 4.4 illustrates the steps involved in the discovery and identification of TA and translation of TA into clinical trials. It is often the case that phase I and II clinical trials demonstrate a degree of efficacy, but in randomized phase III clinical trials, patients failed to demonstrate statistically significant survival benefit [38]. Sipuleucel-T (Provenge), developed by Dendreon corp., is the first vaccine therapy to gain FDA approval and relies on programming dendritic cells (DC) in vitro against recombinant PAP protein. This approach demonstrated an overall extended life expectancy, but no significant effect on time to progression [39]. Results of this trial did not identify the peptide potentially targeted by this treatment. Although results are encouraging, there is still a need to improve the design of vaccination based on a better understanding of the mechanisms which promote and sub verse T-lymphocyte responses. DC-based vaccines are also relatively expensive and at present have questionable cost–benefit advantages.
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy




Related posts:
Novel Strategy of Cancer Immunotherapy: Spiraling Up
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Toll-Like Receptor Pathway and its Targeting in Treatment of Cancers
Photodynamic Therapy and Antitumor Immune Response
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
