Fig. 15.1
VEGF receptors: The isoforms are VEGF-A, VEGF-B, VEGF-C, VEGF-D, and PlGF. VEGF A binds to both VEGFR-1 and VEGFR-2. VEGF-B and PIGF (placenta-derived growth factor) bind to VEGFR-1. VEGF-C and VEGF-D bind to VEGFR-3 and VEGFR-2. B: VEGF and PGF can also bind to heterodimeric receptor complexes formed by VEGFR-1 and VEGFR-2. Likewise, VEGF-C and VEGF-D are known to bind heterodimers formed by VEGFR-2 and VEGFR-3
Table 15.1
Anti-angiogenesis inhibitors, their intracellular targets, and indications
Name | Drug target | Indication(s) | Status |
|---|---|---|---|
Bevacizumab (Avastin®) | Monoclonal antibody for VEGF | FDA approved | |
Sorafenib (Nexavar®), | Multi kinase inhibitor (KIT, FLT-3, RET, VEGFR-1, VEGFR-2, VEGFR-3, and PDGFR-ß) | Metastatic colorectal cancer (mCRC), metastatic renal cell carcinoma (mRCC), non-small cell lung cancer (NSLC), and glioblastoma | FDA approved |
Sunitinib (Sutent®) | Multikinase inhibitor (PDGFRα, PDGFRβ), VEGFR1, VEGFR2, VEGFR3, stem cell factor receptor (KIT), Fms-like tyrosine kinase-3 (FLT3), colony-stimulating factor receptor type 1 (CSF-1R), and the glial cell-line-derived neurotrophic factor receptor (RET) | Gastrointestinal stromal tumor (GIST), renal cell carcinoma (RCC), and advanced pancreatic neuroendocrine tumors (sPNET) | FDA approved |
Pazopanib (Votrient®) | Multi-tyrosine kinase inhibitor VEGFR-1, VEGFR-2, VEGFR-3, PDGFR-α and PDGFR-β, FGFR-1 and FGFR-3, cytokine receptor (Kit), interleukin-2 receptor, inducible T-cell kinase (Itk), leukocyte-specific protein tyrosine kinase (Lck), and transmembrane glycoprotein receptor tyrosine kinase (c-Fms) | Renal cell carcinoma (RCC) and soft tissue sarcoma | FDA approved |
Axitinib (Inlyta®) | VEGFR-1, VEGFR-2, and VEGFR-3 | Renal cell carcinoma (RCC) | FDA approved |
Cabozantinib (Cometriq®) | RET; MET; VEGFR-1, VEGFR-2, and VEGFR-3; KIT; TRKB; FLT-3; AXL; and TIE-2 | Metastatic thyroid cancer (MTC) | FDA approved |
Vandetanib (Caprelsa®) | EGFR and VEGFR families, RET, BRK, TIE2, and members of the EPH receptor and Src kinase families | Thyroid cancer | FDA approved |
Semaxanib | VEGFR2 | None | Discontinued |
Ramucirumab (Cyramza™) | VEGFR2 antagonist that specifically blocks binding of VEGFR ligands, VEGF-A, VEGF-C, and VEGF-D | Gastric adenocarcinoma and esophageal adenocarcinoma | FDA approved |
Everolimus (Afinitor®) | mTOR | Breast cancer (BC) and neuroendocrine tumors | FDA approved |
In addition to the abovementioned compounds, some other low molecular weight VEGFR antagonists such as 5-{3-[4-(octadecyloxy) phenyl] propionylamino}-2,4′-oxydibenzoic acid (VGA1102) and 5-[N-methyl-N-(4 octadecyloxyphenyl)acetyl]amino-2-ethylthiobenzoic acid (VGA1155) were also found to prevent angiogenesis by binding to VEGFR1 and VEGFR2 in NIH3T3 cells (Ueda et al. 2003). The abovementioned VGA1102 and VGA1155, referred as VGA compounds, were expected to be good inhibitors for VEGFR1 (flt-1) and VEGFR2 (KDR/flk-1) while they were not inhibiting the binding of other ligands to their receptors, such as EGF, PDGF, IL-8, PAF (platelet activating factor), IL-1b, IL-2, IL-4, IL-6, MIPs, TNF-a, and insulin. One of the unusual features of these small molecules is that both of them have a long hydrophobic chain (−C18H37; which is referred as the “hydrophobic tail” in this chapter). Even though the VGA compounds are specific to VEGFRs, the actual features of the binding sites of the VEGFR1 are yet to be fully defined. Also, until recently, no inhibitors were reported for the extracellular domain VEGFRs that can effectively block angiogenesis. Therefore, based on the structures of known VEGFR kinase inhibitors, a series of basic chemical scaffolds were built and a similarity search of several databases was carried out. The compounds that emerged from this search were analyzed for their angiogenesis-inhibiting ability using in vitro angiogenesis assay and subsequently by using in vivo tumor model.
15.2 Molecular Modeling
The VEGF inhibitors that are discussed in this chapter were originally identified using the available crystal structure of the apo VEGFR2/KDR molecule which had several missing residues (Sridhar et al. 2005). Therefore, a homology model was generated using the tyrosine kinase domain of fibroblast growth factor receptor 1 (FGFR1) that complexes with an ATP inhibitor SU4984 as a template structure (Mohammadi et al. 1997). The missing loops of the VEGFR2/KDR were built using the “loop model” building option that was available in the Modeller-6v2 program in order to complete the receptor structure (Fiser et al. 2000). The receptor model was refined by energy minimization routine, and also the quality of the refined model was checked with all necessary criteria. To define the pharmacophores for suitable ligand binding, the acceptor (A) and donor (D) sites of the Flex pharmacophores were defined with appropriate distance constraints by measuring from the backbone residues Cys919 and Glu917 of the VEGFR2 protein kinase (Fig. 15.2). For the initial identification and confirmation of the binding sites, the structural features of the VGA compounds were used. Typically the VGA compounds contain hydrophobic tail on one side and the two rings connected by the linker atoms (head) on the other side (Fig. 15.3). It was assumed that the compounds with the hydrophobic tail could bind at the interface between VEGF and its receptor and thereby disturb the critical water-mediated interactions important for the recognition of VEGF to its receptor. The hydrophobic patches on the surface of the VEGFR created for the molecular modeling revealed three hydrophobic sites that could accommodate the hydrophobic tail of the VGA compounds. The VGA compounds were manually docked into these three binding sites, and the docked complexes were energy minimized. The binding pocket contained two parts: Site-A, which was a shallow pocket consisting of hydrophilic/hydrophobic residues, and Site-B that is surrounded by the hydrophobic residues (Fig. 15.4) that extend toward the dimer interface. Interestingly, the identified binding pocket matched the pharmacophores (head and tail side) of the VGA compounds.
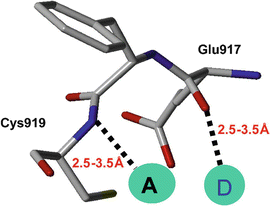
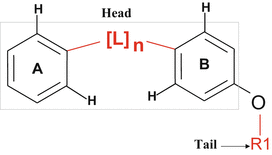
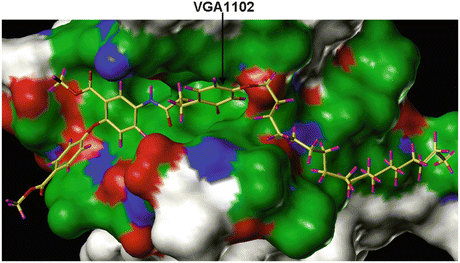
Fig. 15.2
Backbone residues defined as pharmacophores, depicting the two residues (Glu917 and Cys919) with which the ligands interact by making hydrogen bonds. A is the receptor atom from ligand and D is the donor. The hydrogen bond distances are shown by dashed lines, and the distance ranges utilized for database search are also shown
Fig. 15.3
The structure of the VGA compound that contains hydrophobic tail on one side and the two rings connected by the linker atoms (head) on the other side. It was assumed that the compounds with the hydrophobic tail (R1) could bind at the interface between VEGF and its receptor and thereby disturb the critical water-mediated interactions important for the receptor activation
Fig. 15.4
The crystal structure of the VEGFR2/KDR molecule showing the binding of VGA compound that contains hydrophobic tail on one side and the two rings connected by the linker atoms (head) on the other side. The model shows that the compounds with the hydrophobic tail could bind at the interface between VEGF and its receptor and thereby disturb the receptor activation
Using the above indicated simple pharmacophore model, a database that contained 18,000 structures was searched using the Unity Search. At the end a total of 769 compounds were identified from the database that met the pharmacophore requirements as specified in Fig. 15.2. Accordingly, it was anticipated that compounds satisfying those pharmacophoric points will have a high probability of being biologically and structurally diverse. Since in vitro screening of 769 compounds using a low-throughput system is quite time-consuming, it was decided to further reduce the number of initial hits to make the screening process more efficient. Since the goal was to identify good inhibitory compound from the previous dataset of 769 structures, the search was further refined using the UNITY 2D similarity selection strategy using the isoindolinone moiety. As a result of the abovementioned refinement, finally 34 hits (ligands) were obtained by docking to VEGFR with the necessary geometric optimizations. The 34 compounds selected from the refined search were tested for anti-angiogenic ability through in vitro and in vivo assays.
15.2.1 In Vitro Angiogenesis Assay
One of the most common in vitro angiogenesis assays used for screening candidate compounds for inhibiting VEGFR-mediated signaling is done with the human umbilical vein endothelial (HUVEC) cells. These cells are normally maintained in EGM (endothelial growth medium) supplemented with 2 % FBS, 0.1 % EGF, 0.1 % hydrocortisone, 0.1 % GA-1000, and 0.4 % BBE.
Morphogenesis assay on Matrigel is performed using the ECMatrixTM that consists of laminin, collagen type IV, heparan sulfate, proteoglycans, entactin, and nidogen. The ECMatrixTM composition and incubation condition can be optimized for different batches of HUVEC cells for maximal tube-formation in 96-well plates. During the actual assay, the ECMatrixTM-coated 96-well plates are incubated at 37 °C for 1 h to allow the Matrix solution to solidify. While the gel is solidifying, the HUVECs that are grown for 24 h in EGM with 2 % FBS can be trypsinized and resuspended in the growth media for plating. After 1 h pre-incubation of the plate with Matrix solution, the HUVECs are typically plated at a density of 104 cells/well in the absence or in the presence of selected VEGFR inhibitors (concentration range of 0.01–10 μM) that were going to be screened. After 8 h of incubation at 37 °C, the three-dimensional organization (cellular network structures) can be examined under an inverted microscope. When the HUVEC cells are growing on the gel matrix, activated endothelial cells easily form cellular networks (mesh-like structures) resembling capillary tubes sprouting into the stromal space (Fig. 15.5). The formation of these cellular networks is a dynamic process, which starts with the migration and alignment of cells followed by the development of capillary tubelike structures, sprouting of new branches, and finally the formation of the networks.
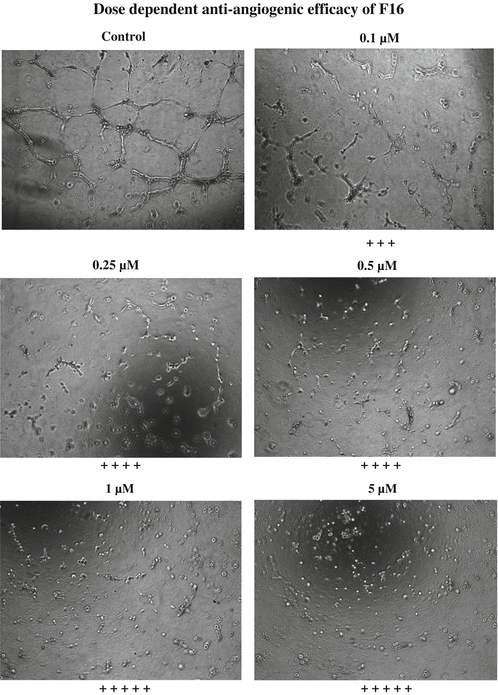
Fig. 15.5
Results of the Matrigel® assay with different concentrations of F16 treatment
Although this in vitro angiogenesis assay is designed only as a qualitative assay, the degree of angiogenesis can be quantitated by using a scoring method as indicated in Table 15.2. The scoring can be based on the extent of the cellular networks as follows: According to this qualitative assessment, the higher the scoring, the greater is the efficiency of the compounds for inhibiting endothelial cell-mediated angiogenesis. The compounds described in this chapter F16 and JFD have greater efficiency in affecting the network formation by the HUVEC cells (Figs. 15.5 and 15.6) and therefore scored four and five pluses at 1 and 10 μM, respectively. These compounds inhibited the angiogenesis to a greater extent, and the individual cells were well separated at both 1 and 10 μM concentration than the lower concentrations. The EC50 values for anti-angiogenic effect of F16 and JFD compounds were estimated to be 0.025 and 0. 25 μM, respectively, as shown in Figs. 15.7 and 15.8. In addition to the angiogenesis inhibition assay, a competitive receptor binding assay was also performed with florescence-labeled VEGF using the HUVEC cells. The lead compounds F16 and JFD were able to displace VEGF binding which indicated its ability to interfere with the binding of VEGF to the receptor.
Table 15.2
 Prevalence of Gastrointestinal Cancers in India
Prevalence of Gastrointestinal Cancers in India
 Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
 TG2: Player That Dictates the Rules in Cancer Progression
TG2: Player That Dictates the Rules in Cancer Progression
 Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
 Current Treatment for Gallbladder Cancer
Current Treatment for Gallbladder Cancer
 Role of Surfactants in Regulation of Cancer Growth
Role of Surfactants in Regulation of Cancer Growth




A scoring system for the assessment of anti-angiogenic effect in the EC Matrix™ assay
Related posts:
Prevalence of Gastrointestinal Cancers in India
Role of Osteopontin in Tumor Microenvironment: A New Paradigm in Cancer Therapy
TG2: Player That Dictates the Rules in Cancer Progression
Seizing Cancer Completely Through Specific Ablating Cancer Stem Cell: The Royal Road to Chemoquiescence
Current Treatment for Gallbladder Cancer
Role of Surfactants in Regulation of Cancer Growth
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
