Figs. 13.1 and 13.2
Antiphospholipid antibody-associated skin ulcers
Expert Opinion 1 (Paulo Ricardo Criado, Dermatology)
Livedoid vasculopathy is a recurrent painful ulcerative disorder, on the skin of the lower extremities, associated with coagulation disorders, autoimmune connective tissue diseases, paraproteinemia, and neoplasia [23, 24]. It results from dermal capillary and small-vessel thrombosis and/or insufficient fibrinolysis due to endothelial, platelet, or coagulation dysfunction [3].
Dermatological manifestations of APS are classified as thrombotic or non-thrombotic. Thrombotic manifestations present as necrotic ulcers from different diseases like LV, Degos disease, pyoderma gangrenosum-like ulcers, necrotizing purpura, thrombophlebitis, periungual ulcerations, multiple linear subungual hemorrhages, digital gangrene and disseminated superficial cutaneous necrosis, and purpura. Non-thrombotic manifestations include livedo reticularis, livedo racemosa, acrocyanosis, primary anetoderma, blue finger syndrome, chronic pigmentous purpura, and chronic urticaria [25, 26].
The diagnosis of LV is made by a deep skin biopsy that includes the epidermis, dermis, and hypodermis, since cutaneous polyarteritis nodosa (cPAN) must be excluded. (In cPAN subcutaneous leukocytoclastic arteritis may be missed in a superficial skin biopsy.)
The treatment of LV is heterogeneous. There are no prospective and controlled studies, and all agents or interventions used are off-label. Our dermatology study group in Brazil proposes an individualized sequential step-by-step treatment (Table 13.1) and is based on the cost-effectiveness and risk-benefit options.
Table 13.1
Proposed step-by-step management algorithm for livedoid vasculopathy
Step 1 | Stop smoking or nicotine patches Treat the underlining thrombophilia (if APS: acetylsalicylic acid / anticoagulation) Treat venous stasis [diosmin + hesperidin (flavonoids mixture with phlebotonic properties) + pentoxifylline and stockings if not contraindicated + vascular surgery referral] [27] Use analgesic drugs (such as tramadol, gabapentin, or pregabalin) [28] |
Step 2 | |
Step 3 | |
Step 4 |
In conclusion, LV is a dermatological manifestation of APS; a correct clinical and histopathological diagnosis is necessary to optimize treatment. Many off-label drugs are used to treat this condition. Controlled studies will be necessary [32].
Expert Opinion 2 (Kurosh Parsi, Dermatology)
The patient presented here seems to have been appropriately diagnosed with LV; however, the diagnosis needs to be confirmed by a skin biopsy. Vascular studies, including venous incompetence studies, should also be performed to exclude coexistent venous hypertension. Differential diagnosis includes venous ulcers, arterial ulcers, lupus panniculitis, pyoderma gangrenosum, skin neoplasms such as squamous cell carcinoma, embolic events, and deep fungal or mycobacterial infections [1, 2].
Treatment of LV is challenging and may require treatment of both the associated coagulopathy and venous hypertension. General measures include avoiding temperature variations that trigger the ulceration. The associated venous hypertension gets worse in heat while vasospasm of microvessels gets worse in cold. Hence both extremes of temperatures result in worsening of the condition [1, 2].
Chronic venous insufficiency results in stagnation of blood flow in the superficial venous network and a predisposition to thrombosis. Improved outcomes have been reported with compression therapy. The best grade of compression is class II (20–30 mmHg), unless contraindicated due to associated peripheral arterial disease. Ankle brachial index measurements and arterial duplex studies may be required in high-risk patients. Treatment for venous disease, using a combination of intravenous laser ablation and foam sclerotherapy, may heal a patient’s ulcers and clear skin pigmentation. Treatment of associated venous hypertension may expedite the healing of ulcers and almost complete clearance of the associated pigmentation (manuscript in press).
Many of the documented treatments for LV focus on anticoagulant therapy with warfarin, heparin, or low-molecular-weight heparin (LMWH) and rivaroxaban [13, 14]. The evidence is mostly anecdotal and based on small case series. Antiplatelet agents , all used off-label, and which include aspirin, clopidogrel, ticlopidine, pentoxifylline, and dipyridamole, have been tried with varying success [1, 2, 10].
The patient in the case has been refractory to anticoagulation, which does not heal ulcers. She should use class II compression and she should be assessed for venous disease, which, if present, should be treated with laser ablation and foam sclerotherapy. The latter is useful to ablate abnormal vessels in the region of ulceration.
Oral anti-inflammatory drugs, such as oral corticosteroids and non-steroidal anti-inflammatory drugs, and antineutrophilic agents such as colchicine, have a role in the management of patients with an underlying inflammatory disease. Other proposed treatments, based on anecdotal experience, include nicotinic acid, hyperbaric oxygen, calcium channel blockers, L-arginine, IVIG, and danazol. Severe cases have responded to tissue plasminogen activator and to intravenous immunoglobulin [1, 2, 17, 18], all off-label uses.
Livedoid vasculopathy is relatively easy to diagnose but challenging to manage. Treatment should reduce the thrombotic load in the dermal microvasculature. Affected patients should be referred to a specialist, such as a vascular physician or a vascular dermatologist. Venous hypertension should be actively treated.
Severe Thrombocytopenia
Literature Review
Thrombocytopenia is the most common hematological manifestation of APS, with a frequency ranging from 20% to 50%. The differences in prevalence reported in studies depend mostly on different threshold descriptions of thrombocytopenia. In most cases, thrombocytopenia is mild (50–150 × 109/L); severe thrombocytopenia (<20 × 109/L) is rare. Even in the latter cases, hemorrhage is far less common than thrombosis [33, 34].
In a retrospective study of 44 thrombocytopenic patients with aPL, bleeding did not occur, and 14 (32%) had thrombotic events. In an Italian registry of aPL, although 25% of 319 patients with APS had thrombocytopenia, only four suffered severe bleeding [34]. Similarly, in 32 patients with severe thrombocytopenia (<50 × 109/L), three had thromboses and two had hemorrhage [35].
The pathogenesis of thrombocytopenia in APS is potentially multifactorial [36]. In animal models and in vitro studies, aPL binds and activates platelets; thus, an aPL-mediated platelet destruction may contribute to thrombocytopenia in APS patients. On the other hand, severe thrombocytopenia correlates more closely with antiplatelet glycoprotein (GP) antibodies than it does with aPL. Antibodies directed against platelet surface GPs have been identified in 40–70% of thrombocytopenic patients with APS, similar to what is seen in idiopathic thrombocytopenic purpura (ITP) [37, 38].
Because there are no guidelines for treatment of APS-associated thrombocytopenia, the ITP guideline is used as a reference [39]. Treatment is necessary in cases of severe thrombocytopenia (<20 × 109/L) or of bleeding. Glucocorticoids, IVIG, immunosuppressive therapies (azathioprine, cyclophosphamide), danazol, and hydroxychloroquine are possible and effective therapies [36, 40–43]. Rituximab may be an alternative in refractory thrombocytopenia. In a literature review, 30% cases treated with rituximab had complete response and 40% a partial improvement of platelet counts [43]. Splenectomy is an option for refractory cases; however, because of surgery-associated thromboses, this should be considered with extreme caution in APS patients [44, 45].
Although some case reports state that eculizumab, a terminal complement inhibitor, is effective for treating catastrophic APS (CAPS) and thrombotic microangiopathy in APS patients [46, 47], there are no data to support its use in other forms of APS-related thrombocytopenia.
Eltrombopag and romiplostim are thrombopoietin receptor agonists, approved for management of chronic ITP. Despite their efficiency in rapidly raising platelet levels, recent case reports have showed severe thrombotic events (including CAPS, and deaths) after its administration in patients with aPL [48–50]. We believe that treatment with thrombopoietin receptor agonists should not be used in patients with APS patients.
Before introducing anticoagulants, platelet levels should be higher than >50 × 109/L [35]. A difficult dilemma is to diagnosis a new thrombotic event in an APS patient with severe thrombocytopenia <30 × 109/L). Failure to address the clot may be life-threatening, while anticoagulation may lead to hemorrhagic complications. Low-dose anticoagulation with unfractionated heparin associated with immunosuppressant may be an option [51].
Case Presentation
A 34-year-old woman, previously healthy, was admitted because of severe chest pain. The electrocardiogram revealed ST elevation, and she was promptly taken to coronary catheterization. The coronary study showed thrombi in the circumflex coronary and excluded atherosclerotic coronary disease. The thrombus was aspirated; no stent was placed. She received anticoagulation and antiplatelet drugs and was sent to the intensive care unit. Surprisingly, the first laboratory results from the emergency room showed a platelet count of 7 × 109/L. Further investigation revealed a strongly positive LA and high titer anticardiolipin antibody (aCL) IgG. Anti-β2-glycoprotein-I antibody (aβ2GPI) was negative. The rheumatology team decided to discontinue antiplatelet drugs and keep the unfractionated heparin, with a strict control. She also received methylprednisolone (1 mg/kg) and IVIG with an improvement in platelet levels to 85 × 109/L. The patient was discharged with stable platelet levels (around 80 × 109/L) on warfarin, prednisone, and hydroxychloroquine. During hospitalization, virus infection, thrombotic thrombocytopenic purpura, bone marrow disorders, systemic lupus erythematosus, and other collagen diseases were excluded. After 3 months, repeat aPL tests confirmed the double positivity and the APS diagnosis. This patient is now taking only warfarin and hydroxychloroquine, has a higher platelet count, and has had no recurrence of thrombosis.
Expert Opinion (Reyhan Diz-Kucukkaya, Hematology)
Platelets are the major cellular component of a thrombus, especially in arterial thrombosis; development of thrombosis in a patient with severe thrombocytopenia is rare. Antiphospholipid syndrome, hemolytic uremic syndrome, thrombotic thrombocytopenic purpura, disseminated intravascular coagulation, heparin-induced thrombocytopenia-thrombosis, and acute leukemia are possible causes [52]. Interestingly, patients with ITP have an increased risk of thrombosis, but, even in severe thrombocytopenia, hemorrhage is far less common than is thrombosis, as in the present case. Several explanations have been postulated for the development of thrombosis in patients with ITP including the presence of aPL itself, activation of the complement system, and individual risk factors for thrombosis [36, 53, 54].
Immunosuppressive therapies may decrease titers of antiplatelet antibodies and increase platelet counts in APS patients with thrombocytopenia, but they do not reduce the titers of aPL [55], suggesting that thrombocytopenia is a secondary phenomenon in at least some APS patients. Thrombotic microangiopathy may contribute to both thrombocytopenia and thrombosis in a subset [56].
Severe thrombocytopenia in patients with APS is treated similarly to that in patients with ITP; it usually responds well to glucocorticoids and immunosuppressive drugs. Although IVIG may increase the platelet count very rapidly in these patients, the thrombotic risk of IVIG itself should be considered [57]. The data concerning the use of thrombopoietin receptor agonists in patients with APS is limited to case reports and was associated with an increased risk of thrombosis [50, 58].
The choice and monitoring of anticoagulant therapy in APS patients with severe thrombocytopenia and thrombosis are also challenging. Although unfractionated heparin is preferred in patients with acute coronary syndrome, activated thromboplastin time and activated clotting time are prolonged in patients having LA; thus, anti-factor Xa and protamine titration assays are recommended for heparin monitoring. The use of low-molecular-weight heparin is appealing in treating patients with LA, since it causes a more predictable anticoagulant effect [59].
Although most patients with aPL and thrombocytopenia will not require treatment, some, as in this case, represent a treatment challenge. Corticosteroids and immunosuppressants should be considered when clinically significant thrombocytopenia occurs.
Cardiac Valve Disease
Literature Review
Cardiac valve disease (CVD), a non-criteria manifestation of APS, is defined as (a) valve thickness >3 mm, (b) localized thickening involving the proximal or middle portion of the leaflets, or (c) irregular nodules on the atrial face of the mitral valve and/or the vascular face of the aortic valve [60]. Although most of the cases are asymptomatic [61], about 5% of patients with valve disease will progress to cardiac failure, requiring valve replacement [60]. Additionally, a meta-analysis showed that 48% of aPL-positive lupus patients have valve disease compared with only 21% of aPL-negative SLE patients [62].
Valve lesions are associated with high risk of arterial events in primary APS patients [63]. A meta-analysis of 23 studies, including 1656 patients with SLE and 508 cases of valve disease, showed a threefold higher frequency for any valve lesion in SLE patients with aPL, compared to those without aPL. The risk associated with IgG aCL was as high as for LA [64].
On histopathology, these lesions are characterized by superficial and intravalvular deposits of fibrin with subsequent organization [63]. A mechanism implicated in the generation of valve lesions is that an immune-mediated endothelial activation by aPL triggers an inflammatory cascade, resulting in complement deposition and valve damage [60].
According to the 13th International Congress on Antiphospholipid Antibodies Task Force on CAPS and Non-criteria APS Manifestations, transthoracic echocardiogram (TTE) in APS patients with thrombosis (mainly arterial) is recommended. In patients with normal valves and in the absence of atherosclerotic risk factors, follow-up may be unnecessary. If valve lesions exist, serial echocardiographic follow-up is suggested [65]. According to the American College of Cardiology, transesophageal echocardiogram (TEE) should follow a non-diagnostic (transthoracic echocardiogram) TTE. However, TEE may be the initial test in patients with a suspected cardioembolic event and no history of atrial fibrillation [66]. Two-dimensional echocardiography studies are the standard modality for diagnosis [67, 68]. Transesophageal echocardiogram is more accurate for detection of vegetations and thickening compared to Transesophageal echocardiogram (73% vs. 39%) [69].
A study comparing different techniques for cardiac assessment in APS concluded that cardiovascular magnetic resonance (CMR) identifies a high prevalence of occult myocardial scarring and endomyocardial fibrosis in APS. If quantification of heart valve disease and stress myocardial perfusion-fibrosis is needed, CMR is the technique of choice [70].
The optimal treatment of aPL-related cardiac valve disease is unknown. Oral anticoagulant treatment with an INR goal between 2.0 and 3.0 and aspirin (100 mg/day) was not effective for valve lesion regression [71]. Corticosteroid treatment was not effective for improving valve healing [72]. Valve replacement in patients with APS carries significant early and late morbidity and mortality [73]. Surgical risk is even greater when active SLE and renal involvement are present [74]. The successful use of transcatheter aortic valve replacement (TAVR) was reported in a single case study of the treatment of severe aortic stenosis in a patient with active SLE and APS [75].
Case Presentations
Case 1
A 29-year-old female, taking an oral contraceptive, with a history of migraine and premature delivery at 34-week gestational age, presented with left-sided facial numbness due to a right thalamic stroke while. She was triple positive for aPL. She was started on LMWH and bridged to warfarin. Two weeks later, brain magnetic resonance imaging (MRI) revealed multiple scattered, bilateral supratentorial and infratentorial acute infarctions in the setting of a subtherapeutic INR; therefore, LMWH was reinitiated. Two days later, she reported an episode of aphasia for several hours. A new brain MRI showed multiple new foci of diffusion restriction consistent with acute infarction. Transthoracic echocardiogram was normal but TEE showed a sessile echo dense lesion (0.4 × 0.2 cm) on the left atrial surface of the anterior mitral leaflet (Fig. 13.3).
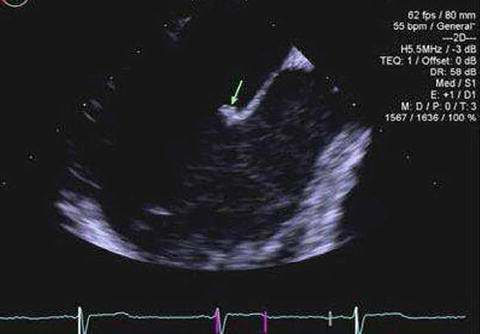
Fig. 13.3
Antiphospholipid antibody-associated cardiac valve disease; a small, sessile echodensity on the left atrial surface of the anterior mitral leaflet (Permission to publish was obtained from Arthritis Care & Rheumatology)
Case 2
A 38-year-old female, taking an oral contraceptive, with no prior history of thrombosis, presented with cyanosis of the left second finger. Ultrasonography and magnatic resonance angiography were normal; she was found to have triple aPL positivity and was started on LMWH with quick improvement of the pain and color of fingers. Transthoracic echocardiogram was normal, but TEE showed a small mobile echo-dense lesion on the aortic valve. Both patients had persistent aPL titers when repeated in 12 weeks.
Expert Opinion (Mary Carmen Amigo, Rheumatology)
Cardiovascular disease in APS patients is associated with stroke, transient ischemic attack (TIA), epilepsy, and migraine [76]. Heart failure, infective endocarditis, valve replacement, and death are all complications of valve damage irrespective of its etiology. Lupus anticoagulant positivity with mitral thickening/regurgitation is associated with a tenfold greater risk of cerebral infarcts in patients with lupus [77]. In APS, CVD is associated with an 8.4-fold risk of arterial thrombosis, as reported in a 12-year follow-up study [78].
Case 1 has APS with arterial thrombosis and a high risk of re-thrombosis. RATIO study showed that the odds ratio for ischemic stroke in women with positive LA was 43.1 (12.2–152.0) but increased to 201.0 (22.1–1828.0) if the women was taking an oral contraceptive [79, 80]. In the second case, as there is no confirmation of thrombosis or pregnancy morbidity, a diagnosis of APS based on classification criteria [81] is not possible. However, digital ischemia and an aortic valve vegetation with triple aPL positivity strongly suggest that she is a high-risk patient.
In patients with a high-risk profile, aspirin is not effective for primary thromboprophylaxis and is significantly less effective than vitamin K antagonists for secondary prevention [82, 83]. There is no consensus regarding optimal antithrombotic management of patients with ischemic stroke/TIA and aPL (independent of valve disease). We look forward to data from ongoing studies on the efficacy and safety of the new oral anticoagulants in patients with APS.
In case 1, a long-term oral anticoagulation (INR > 3.0) or long-term oral anticoagulation (INR 2.0–3.0) with low-dose aspirin (100 mg/day) is the best approach [84–86]. Some investigators suggest that D-dimer level is an indicator of a higher susceptibility to embolism recurrence [87]. Literature on atrial fibrillation patients demonstrates that patients with elevated D-dimer during oral anticoagulation therapy are at high risk for thromboembolic events [88].
In case 2, as digital ischemia is a potentially serious complication, it requires a prompt assessment and introduction of treatment. The initiation of LMWH promptly improved pain and color of the fingers. Treatment alternatives include LMWH, warfarin (INR > 3), or warfarin (INR 2.0–3.0) plus low-dose aspirin [84].
Even though there is no evidence-based data, HCQ or statins can be considered as additional treatments [43]. In a small cohort of refractory cases, IVIG proved to be beneficial [89]. Cognitive dysfunction, a common and serious complication in patients with cerebral ischemia, merits consideration of rituximab, which may benefit non-criteria manifestations of APS such as thrombocytopenia, skin, and valve disease [22]. Scant data regarding corticosteroids for aPL-associated valve disease suggest they are not effective [90, 91].
Antiphospholipid Antibody-Associated Nephropathy
Literature Review
Antiphospholipid antibody-associated nephropathy is a non-criteria APS manifestation [81]. This small-vessel nephropathy, called APS- or aPL-associated nephropathy, was first described in primary APS [95] and further described in SLE patients with positive aPL, with and without APS [96].
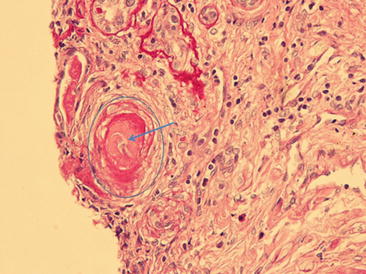
Antiphospholipid antibody-associated nephropathy is characterized by thrombotic microangiopathy (the acute lesion) and chronic vaso-occlusive lesions, such as fibrous intimal hyperplasia, organizing thrombi and/or fibrous occlusions of arteries or arterioles and focal cortical atrophy. Other causes of renal microangiopathy such as malignant hypertension, diabetes mellitus, thrombotic thrombocytopenic purpura, and systemic sclerosis [65, 81] should be excluded.
The major clinical characteristics of this nephropathy include mild-to-severe hypertension, microscopic hematuria, proteinuria (mild to nephrotic level), and renal insufficiency. The latter is usually mild but may progress to renal failure [97].
The 13th International Congress on Antiphospholipid Antibodies Task Force on Non-criteria APS Manifestations critically evaluated studies on the relationship between aPL and aPL-associated nephropathy [65] and concluded that, among primary APS patients, aPL-associated nephropathy accounted for 90–100% of all biopsy-proven renal involvement and for 67–100% of patients with SLE-APS who had renal involvement [65, 96].
There is no consensus on the management of aPL-associated nephropathy patients, which may occur despite full-dose anticoagulation and may not improve if anticoagulation/antiplatelet therapy is initiated after diagnosis. Empirical options are primarily focused on the management of hypertension and proteinuria [98]. Based on case reports or small case series, angiotensin-converting enzyme (ACE) inhibitors , aspirin, oral anticoagulants, hydroxychloroquine, corticosteroids, and/or immunosuppressive agents can be alternatives [99–101].
In patients with aPL but without APS, aspirin and/or hydroxychloroquine can be considered, especially in patients with SLE. Oral anticoagulants may be used in patients with high-risk aPL profile, such as those with persistently positive LA, persistently positive aCL in medium-high titers, and those with triple positivity [102]. The control of blood pressure and proteinuria, using mainly ACE inhibitors, is recommended for all the patients with aPL-associated nephropathy [96]. Other treatment options for more refractory cases, based on few studies, include IVIG [89], rituximab [22], and eculizumab [85, 103].
Case Presentation
A 21-year-old woman with APS (triple positive aPL, prior dural venous sinus thrombosis requiring ventriculo-peritoneal shunt, DVT extending to the right common and deep femoral veins, chronic occlusion of the celiac and right external iliac artery, and ulcer on the left tibia) developed moderately elevated blood pressure. Laboratory tests confirmed persistent proteinuria of 1.8 g/24 h and active urine sediment with normal serum creatinine levels. The patient’s left lower extremity ulcer worsened with painful superficial ulcerations of the skin bilaterally and development of necrosis requiring surgical debridement. A repeat skin biopsy was consistent with small-vessel vasculopathy. Renal biopsy demonstrated aPL-associated nephropathy lesions (Fig. 13.4). Treatment with rituximab was initiated, with slowly decreasing 24-h urinary protein levels, stable renal function, and gradual improvement and ultimate healing of the leg ulcers.
 Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
 Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
 Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis




Related posts:
Recent Advances in Understanding of the Genetics of Antiphospholipid Syndrome
Clinical and Prognostic Significance of Non-criteria Antiphospholipid Antibody Tests
Prevention and Treatment of Obstetric Antiphospholipid Syndrome
15th International Congress on Antiphospholipid Antibodies Task Force on Catastrophic Antiphospholipid Syndrome Report
Mechanisms of Antiphospholipid Antibody-Mediated Pregnancy Morbidity
Mechanisms of Antiphospholipid Antibody-Mediated Thrombosis
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
