Recent breakthroughs in the fundamental understanding of the cellular and molecular basis of melanoma have culminated in new therapies with unquestionable efficacy. Immunotherapy and targeted therapy strategies have completely transformed the contemporary management of advanced melanoma. The translational research behind these developments is discussed, with an emphasis on immune checkpoint blockade and inhibition of the mitogen-activated protein kinase signaling pathway.
Key points
- •
Recent advances in systemic therapy for melanoma have altered the outlook for patients afflicted with advanced stages of the disease.
- •
Effective immunotherapy is associated with infrequent and slowly developing responses, which may result in durable remission of disease at the expense of significant autoimmune toxicities.
- •
Targeted therapies are associated with high response rates and rapid tumor regression, however drug resistance almost invariably develops.
- •
Robust biomarkers need to be identified in order to increase the probability that the individual patient is likely to benefit from a specific therapeutic strategy.
Melanoma is the fifth most common malignancy in the United States, representing a remarkable increase in incidence, as noted in population studies. It follows that melanoma also carries one of the fastest increasing death rates. Surgery has long been the cornerstone of melanoma therapy, with only limited efficacy reported from an array of adjuvant strategies and therapies for unresectable disease. Although survival for patients with localized disease is excellent, survival declines precipitously for those with metastatic disease, who have an expected 2-year survival rate of 10% to 20%. Until 2011, only 3 drugs had been approved as treatment options for patients with metastatic melanoma: dacarbazine (DTIC), fotemustine, and interleukin 2 (IL-2). Standard chemotherapy regimens have response rates of approximately 15%, and duration of response tends to be on the order of months, although durable, complete responses (CRs) have been reported. Combinations of cytotoxic agents have been investigated and have not substantially improved survival outcomes compared with DTIC alone. Despite vigorous efforts to bring new therapies from the bench to bedside, approval by the US Food and Drug Administration (FDA) of the cytokine IL-2 in 1998 was the only new therapy to arise in more than 30 years after standard cytotoxic chemotherapy regimens had been established. Effective therapies for unresectable melanoma have long represented an unmet need based on the rapidly increasing incidence and poor prognosis of advanced disease states.
During the past 2 years, there has been renewed optimism as the translation of basic scientific findings into positive clinical trial data has resulted in the approval of efficacious new agents. In addition, several promising new therapies are in the final stages of clinical testing. These advances have evolved through 2 seemingly unrelated approaches: immunotherapy and targeted therapy. This review highlights important recent translational research findings and their impact on the care of patients with melanoma ( Table 1 ).
| Reference | Classification | Patients (n) | Treatment | Response Rate (%) | Duration of Response (mo) | Median PFS (mo) | Overall Survival (%) |
|---|---|---|---|---|---|---|---|
| Schwartzentruber et al, 2011 | Immunotherapy (vaccine) phase 3 | 94 | gp100 + IL-2 | 16 | NR | 2.2 | NR |
| 91 | IL-2 | 6 | NR | 1.6 | NR | ||
| Bedikian et al, 2010 | Immunotherapy (vaccine) phase 2 | 127 | Allovectin-7 | 11.8 | 13.8 | 1.6 | NR |
| Senzer et al, 2009 | Immunotherapy (vaccine) phase 2 | 50 | OncoVEX GM-CSF | 26 | NR | NR | 58 at 1 y 52 at 2 y |
| Hodi et al, 2010 | Immunotherapy (checkpoint blockade) phase 3 | 137 | Ipilimumab ± gp100 | 10.9 | NR | 2.8 | 45.6 at 1 y 23.5 at 2 y |
| 136 | gp100 | 1.5 | NR | 2.8 | 25.3 at 1 y 13.7 at 2 y | ||
| Robert et al, 2011 | Immunotherapy (checkpoint blockade) phase 3 | 250 | Ipilimumab + DTIC | 15.2 | NR | 2.8 | 47.3 at 1 y 28.5 at 2 y |
| 252 | DTIC | 10.3 | NR | 2.8 | 36.3 at 1 y 17.9 at 2 y | ||
| Topalian et al, 2012 | Immunotherapy (checkpoint blockade) phase 1 | 94 | Anti-PD-1 Ab | 28 | NR | NR | NR |
| Brahmer et al, 2012 | Immunotherapy (checkpoint blockade) phase 1 | 52 | Anti-PD-L1 Ab | 17.3 | NR | NR | NR |
| Chapman et al, 2011 | Targeted therapy (BRAF inhibition) phase 3 | 337 | Vemurafenib | 48.4 | 6.7 | 5.3 | 84 at 6 mo 56 at 1 y |
| 338 | DTIC | 5.4 | NR | 1.6 | 66 at 6 mo 44 at 1 y | ||
| Hauschild et al, 2012 | Targeted therapy (BRAF inhibition) phase 3 | 187 | Dabrafenib | 49.7 | 5.6 (estimated) | 5.1 | NR |
| 63 | DTIC | 6.3 | NR | 2.7 | NR | ||
| Flaherty et al, 2012 | Targeted therapy (MEK inhibition) phase 3 | 214 | Trametinib | 22 | 5.5 | 4.8 | 81 at 6 mo |
| 108 | DTIC or paclitaxel | 8.3 | NR | 1.5 | 67 at 6 mo | ||
| Flaherty et al, 2012 | Targeted therapy (combined BRAF/MEK inhibition) phase 1/2 | 54 | Dabrafenib plus trametinib | 75.9 | 10.5 | 9.4 | 79 at 1 y |
| 54 | Dabrafenib | 53.7 | 6.1 | 5.8 | 70 at 1 y |
Immunotherapy
The foundation for modern cancer immunotherapy for melanoma was the development of the cytokine IL-2 as a therapeutic agent during the 1980s. IL-2 is a common γ-chain cytokine required for T-cell growth and activation. It has no direct tumoricidal activity; rather, its antitumor activity derives from its influence on the cytotoxic activity of T lymphocytes, which recognize and destroy the tumor. Although objective responses occur in only approximately 15% of patients with melanoma, nearly half of responding patients have a CR. With long-term follow-up, complete responders frequently (>80%) have durable remission, with many of these patients believed to be cured. The sustainability of responses and potential for cure clearly distinguish immunotherapy from other available systemic therapies.
Most of the work in the immunotherapy for melanoma has focused on T-cell–mediated responses. Dendritic cells (DCs) are professional antigen-presenting cells (APCs) that capture antigens and present them to naive T cells in the context of activating costimulatory signals, thereby initiating the immune response. In the absence of costimulation, T cells are induced into a state of unresponsiveness, or anergy. Tumor-associated antigens (TAA) are bound to major histocompatibility complex (MHC) class I molecules and presented on the surface of tumor cells. When activated CD8+ T cells with the appropriate receptor encounter a tumor cell showing the TAA, they are able to perform their cytolytic killing functions on the tumor target and release inflammatory cytokines to activate neighboring immune cells. This summary is inadequate to encompass all of the complexities involved in the regulation of cellular immune responses against tumors, but it provides the framework to understand the work to be discussed.
Vaccines
Melanoma vaccines have been investigated as adjuvant therapy in resected patients with high risk of recurrence as well as in the therapy for metastatic disease. Generically, vaccines consist of tumor antigen(s) in a formulation designed to activate the cellular arm of the immune system. A common approach has been the use of peptide vaccines of variable length containing the sequence of the target TAAs. These peptides are typically administered with an adjuvant, such as incomplete Freund’s adjuvant or bacterial derivatives like bacillus Calmette-Guérin, which stimulate immune responses nonspecifically. Vaccination strategies using viruses or naked DNA encoding TAAs have been investigated. A spectrum of strategies with DC vaccines have been explored. Irradiated autologous or allogeneic tumor preparations have been used as vaccines. Vaccines have been administered in conjunction with immunostimulatory cytokines such as IL-2, IL-12, and granulocyte-macrophage colony-stimulating factor (GM-CSF).
There are several conceptually appealing features of vaccine strategies: vaccines are most often administered in the outpatient setting, with minimal need for specialized care; they are typically associated with minimal side effects; and, if successful, they may lead to lasting tumor immunity. Investigations in this area have been disappointing in their failure to consistently and clearly show clinical efficacy. Klebanoff and colleagues recently examined notable contemporary cancer vaccine trials reported since 2004; overall, only 12 of 309 (3.9%) patients with melanoma had objective responses. In retrospect, this entire body of work highlights our incomplete understanding of the mechanisms required to trigger antitumor immunity. Through these early failures, there have been substantial gains in fundamental knowledge and promise for a path to improving on immunization strategies.
The lack of efficacy in vaccine trials has been attributed to a variety of shortcomings. First, the central role of DCs in successful immunization was not recognized as cancer vaccines were first being developed, and this point has only recently been addressed in newer vaccine studies. It has been recognized that adjuvant agents are crucial for the initiation of immune responses, yet the array of approved adjuvants remains woefully limited and their function poorly understood. Despite the identification of hundreds of TAAs, it remains unclear which are the best targets to reach the end point of clinical efficacy. Also, antigen expression within any given tumor is heterogeneous and the ability of the tumor to show these antigens is often compromised. Another previously underappreciated component of vaccine strategies is the need to overcome mechanisms of immunosuppression mediated by the tumor or present in the tumor microenvironment. Each successive generation of studies of cancer vaccines has incorporated strategies based in the continuously expanding knowledge of immunobiology.
We have only recently reached the important benchmark of a positive phase 3 study of melanoma vaccines, with 2 promising unrelated studies soon to be reported. Schwartzentruber and colleagues recently published the findings of a multicenter, phase 3 study evaluating the combination of the peptide vaccine gp100 with high-dose IL-2 compared with IL-2 alone. The addition of the peptide vaccine significantly improved response rates (16% vs 6%) and progression-free survival (PFS) (2.2 months vs 1.6 months). There was a trend toward improved median survival (17.8 months vs 11.1 months; P = .06). This monumental effort validates the potential of vaccine strategies in the treatment of established metastatic melanoma but also underscores the need for innovation to enhance efficacy.
Allovectin-7 is a novel vaccine for metastatic melanoma that has shown promise in early-phase clinical studies. Downregulation or loss of expression of MHC class I and β 2 microglobulin molecules has been identified as 1 mechanism through which tumor cells may escape immune detection. Allovectin-7 is a plasmid DNA encoding HLA-B7 and β 2 microglobulin. This vaccine is injected directly into tumors and results in the assembly of a complete MHC class I complex on the tumor surface. It is proposed that the vaccine may have activity through several mechanisms, including expression of a foreign antigen (in HLA-B7–negative patients), increased TAA presentation in the context of HLA-B7, and increased surface expression of MHC class I molecules on the tumor surface (because of the increased availability of β 2 microglobulin molecules). In addition, it is possible that unmethylated bacterial CpG motifs in the plasmid backbone may stimulate immune responses, as has been shown in animal models. Objective response rates of 4% to 11% have been reported in phase 2 studies, with some patients undergoing CR. Antitumor responses have been reproducible in multiple phase 2 trials, and all studies document an extremely favorable toxicity profile. A phase 3 clinical trial comparing Allovectin-7 with DTIC or temozolomide has recently completed accrual.
Another promising vaccine is OncoVEX GM-CSF , an oncolytic herpes simplex virus type 1 that has been genetically engineered for tumor-selective replication and enhanced tumoricidal activity. In addition, the virus encodes the immunomodulatory cytokine GM-CSF, which stimulates the proliferation, maturation, and differentiation of DCs. The vaccine is injected intratumorally. Responses are seen both in the injected lesions and at distant sites. It is presumed that tumor lysis at the site of injection releases antigens, which are subsequently presented by DCs, initiating systemic antitumor effects. In a phase 2 study, the objective response rate was 26%, with more than half of patients achieving CR. Subsequently, an eagerly anticipated phase 3 study has completed accrual, with results expected shortly.
Adoptive Cell Transfer Therapy
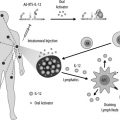
Adoptive cell transfer therapy (ACT) for patients with unresectable melanoma has evolved through elegant basic, translational, and clinical research. ACT involves resection of a melanoma metastasis, isolation of tumor-infiltrating lymphocytes (TILs) from the tumor explant, expansion of the T-cell cultures in vitro, and reinfusion of autologous tumor reactive cells together with IL-2 into the patient, after a lymphodepleting preparation. Transgenic mouse models of ACT have played a major role in the optimization of human protocols. Murine studies have clarified desirable characteristics of transferred T cells, the importance of lymphodepletion, and the role of APCs. The Surgery Branch of the National Cancer Institute recently reported their extensive experience with ACT in the therapy for 93 heavily pretreated patients with metastatic melanoma. Objective response rates greater than 70% were achieved in the most recent series. Durable, complete tumor regression was achieved in 22% of patients, with an actuarial 5-year survival of 93% in patients with CR. No other therapy has matched these outcomes.
More recently, genetically modified peripheral blood lymphocytes have been used in ACT studies. Antitumor activity has been reported in patients with melanoma treated with T cells retrovirally transduced with high-affinity T-cell receptors (TCRs) recognizing tumor antigens such as MART-1, NY-ESO-1, and MAGE-A3. Chimeric antigen receptors (CARs) comprise the tumor antigen-binding domain of an antibody molecule fused to the intracellular signaling domain of a TCR. CARs specific for high-molecular-weight melanoma-associated antigen and ganglioside GD3 have shown promise in preclinical testing. CARs have several theoretic advantages over TCR-based gene transfer. Effector cell engagement does not depend on peptide antigen expression and MHC-restricted presentation by the tumor, potential mechanisms by which tumors may escape immune detection. Because they function in an MHC-independent manner, there may be wider applicability to patients as an off-the-shelf reagent. In addition, there is the ability to engineer these CARs to express the intracellular signaling domains of a variety of costimulatory molecules to improve T-cell function and survival.
Immunomodulation
Clinical trials of IL-2 showed that the immune system could be activated nonspecifically to promote tumor regression in patients with melanoma. One implication of these trials is that patients with melanoma frequently have endogenous lymphocytes capable of recognizing tumor antigens; it has also been established that lymphocytes that recognize the tumor frequently reside near the tumor. Substantial evidence indicates that the tumor interacts with responding lymphocytes to subvert the immune response and permit tumor growth, a process referred to as immunoediting.
According to the classic 2-signal model of T-cell activation, the first signal is provided through the interaction of the antigen-MHC complex with the TCR. Naive T cells encountering an antigen must also receive a second signal to initiate an immune response. This process occurs through the interaction of CD28 receptors on T cells with B7-1 (CD80) and B7-2 (CD86) molecules, which are present on APCs. In the absence of costimulation, T cells become anergic. Subsequently, additional costimulatory pathways providing both activating (CD28, OX40, GITR, CD137, CD27, HVEM) and inhibiting (CTLA-4, PD-1, TIM-3, BTLA, VISTA, LAG-3) second signals have been identified. The interplay of these costimulatory molecules serves to precisely control the intensity and duration of cellular immune responses. Elucidation of these diverse pathways has broadened the concept of costimulation, and these immunologic checkpoints represent new targets for therapeutic intervention.
Cytotoxic T Lymphocyte Antigen 4 Blockade
Cytotoxic T lymphocyte antigen 4 (CTLA-4) was identified as a homologue of CD28, which receives the second signal in T-cell activation. In contrast to CD28, it is not present on the surface of naive T cells but is rapidly upregulated with T-cell activation. CTLA-4 binds B7-1 and B7-2 with significantly higher affinity than CD28, and signaling through CTLA-4 engagement serves to attenuate T-cell activation and proliferation. Its native function has been described a brake that prevents productive immune responses from progressing to autoimmune injury; CTLA-4–deficient mice die at 3 to 4 weeks of age from a fatal lymphoproliferative disorder. CTLA-4 is overexpressed on chronically stimulated and exhausted T cells. In addition, CTLA-4 is constitutively expressed on T-regulatory cells, which may be crucial for their suppressive function. Blockade of CTLA-4 results in enhanced T-cell–mediated immune responses, and numerous animal models have shown that tumor immunity can be induced by interfering with CTLA-4 signaling. Subsequently, ipilimumab, a fully human monoclonal antibody, was developed as a therapeutic agent to block CTLA-4. Early-phase clinical trials reported activity in patients with metastatic melanoma.
Ipilimumab received FDA approval in 2011 after the publication of a phase 3 study in which patients with metastatic melanoma were treated with ipilimumab alone or in combination with the gp100 peptide vaccine. The control arm received only the gp100 vaccine. All patients had received previous therapy with other agents, including more than 20% of patients receiving IL-2. There were significant differences in survival with 1-year and 2-year overall survival of 45.6% and 23.5% in patients receiving ipilimumab compared with 25.3% and 13.7% in patients receiving gp100 alone. Overall survival was independent of previous IL-2 therapy, suggesting that these immunomodulating agents act through different mechanisms. Grade 3 or 4 toxicities were frequent (10% to 15%), and death attributed to immune-related adverse events occurred in approximately 1% of patients. The gp100 vaccine did not enhance the efficacy of CTLA-4 blockade, as was seen when this vaccine was combined with IL-2. This finding reinforces the concept that IL-2 and anti-CTLA-4 have inherently different mechanisms of action. This landmark study is the first randomized clinical trial showing a benefit in overall survival for patients metastatic with melanoma. Another pivotal phase 3 study examined the efficacy and safety of ipilimumab plus DTIC compared with DTIC alone in 502 patients with previously untreated metastatic melanoma. The 1-year, 2-year, and 3-year survival were 47.3%, 28.5%, and 20.8% for patients receiving ipilimumab and DTIC compared with 36.3%, 17.9%, and 12.2% for patients receiving DTIC alone. Grade 3 or higher immune-mediated adverse events were documented in 38.1% of patients receiving ipilimumab; there were no deaths attributed to autoimmune phenomena.
Prieto and colleagues recently reported the longest follow-up on patients with melanoma treated with ipilimumab in 3 consecutive trials conducted at the National Cancer Institute. With median follow-up of 71 to 92 months, this study reveals unique and important features of this therapy that are often not apparent in the randomized studies. First, objective responses frequently evolved slowly; in complete responders, the median time to CR was 30 months, with 1 patient achieving CR at 71 months. Reports with shorter follow-up may underestimate the true objective response rate. Second, CRs are remarkably durable, with only 1 of 15 patients recurring after CR; all of the other patients with CR were alive without evidence of disease with median follow-up of 83 months. This finding is reminiscent of the experience with IL-2, and it is tempting to speculate that some of these patients may be cured.
Programmed Death 1/Programmed Death Ligand 1 Blockade
Programmed death 1 (PD-1) and its ligands, PD-L1 and PD-L2, have recently attracted attention as another immunologic checkpoint that can be manipulated to achieve antitumor immune responses. PD-1 was first identified in 1992 and its ligands nearly 10 years later. Signaling through the PD-1/PD-L1(-L2) axis serves to dampen T-cell–mediated immune responses and plays a critical role in central tolerance induction. Inflammation and cardiomyopathy have been observed in PD-1 knockout mice. This autoimmune phenotype is considerably milder than that seen in CTLA-4 knockout mice and does not manifest until 6 to 9 months after birth. PD-1 is not expressed on naive T cells but is induced quickly on activation. When antigen-specific T cells are chronically stimulated, PD-1 is upregulated, and the cells become functionally impaired; T-cell function can be restored with PD-1 blockade. In TILs derived from patients with melanoma, increased PD-1 expression has been correlated with an exhausted phenotype and impaired effector function. PD-L1 is constitutively expressed in hematopoietic cells and a wide variety of other tissues. Because of this distribution, it has been postulated that this pathway serves to protect normal peripheral tissues from activated T cells, thus limiting autoimmunity. PD-L1 is also expressed by many tumors, including melanoma. In solid tumors, PD-L1 can inhibit the cytolytic activity of tumor-infiltrating T cells. Furthermore, PD-L1 expression has been correlated with an unfavorable prognosis in a variety of cancer histologies. Expression of PD-L1 is believed to be a mechanism by which tumors may escape immune rejection.
Two concurrently published phase 1 clinical trials using monoclonal antibodies to disrupt the PD-1/PD-L1 axis have shown promising clinical efficacy across an array of histologies including non–small cell lung cancer, melanoma, and renal cell cancer. In the study reported by Topalian and colleagues, anti-PD-1 antibody was administered in escalating doses to 296 patients. These individuals included 94 patients with melanoma, for whom the objective response rate was 28%. Most responding patients had durable responses of greater than 1 year. Pretreatment tumor specimens were available from a few study patients, and it was noted that objective responses were observed only in patients whose tumors expressed PD-L1, suggesting that this may be a potential biomarker for patient selection. Grade 3 or higher toxicities occurred in 14% of patients. Potential autoimmune toxicities included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. There were 3 cases of fatal pneumonitis. Brahmer and colleagues reported the companion trial, in which patients with a variety of histologies were treated with escalating does of anti-PD-L1 antibody. In the patients with melanoma, 9 of 52 patients had objective responses (17%), including 3 CRs. As reported with the anti-PD-1 antibody, responses tended to be durable, and stable disease (27%) was a frequent outcome in patients not classified as having an objective response. Grade 3 or 4 toxicities occurred in only 9% of study patients. Potentially immune-related toxicities included rash, hypothyroidism, hepatitis, sarcoidosis, endophthalmitis, diabetes mellitus, and myasthenia gravis. Taken together, these studies show that blockade of PD-1 or PD-L1 can result in tumor regression, with a pattern of response similar to that seen with CTLA-4 blockade. The immune-related toxicities attributed to anti-PD-1 and anti-PD-L1 are less infrequent and milder than those associated with anti-CTLA-4. Phase 2 and 3 trials will further define the efficacy and applicability of these therapies, and FDA approval of 1 or both agents seems imminent based on the dramatic results already reported.
Immunotherapy
The foundation for modern cancer immunotherapy for melanoma was the development of the cytokine IL-2 as a therapeutic agent during the 1980s. IL-2 is a common γ-chain cytokine required for T-cell growth and activation. It has no direct tumoricidal activity; rather, its antitumor activity derives from its influence on the cytotoxic activity of T lymphocytes, which recognize and destroy the tumor. Although objective responses occur in only approximately 15% of patients with melanoma, nearly half of responding patients have a CR. With long-term follow-up, complete responders frequently (>80%) have durable remission, with many of these patients believed to be cured. The sustainability of responses and potential for cure clearly distinguish immunotherapy from other available systemic therapies.
Most of the work in the immunotherapy for melanoma has focused on T-cell–mediated responses. Dendritic cells (DCs) are professional antigen-presenting cells (APCs) that capture antigens and present them to naive T cells in the context of activating costimulatory signals, thereby initiating the immune response. In the absence of costimulation, T cells are induced into a state of unresponsiveness, or anergy. Tumor-associated antigens (TAA) are bound to major histocompatibility complex (MHC) class I molecules and presented on the surface of tumor cells. When activated CD8+ T cells with the appropriate receptor encounter a tumor cell showing the TAA, they are able to perform their cytolytic killing functions on the tumor target and release inflammatory cytokines to activate neighboring immune cells. This summary is inadequate to encompass all of the complexities involved in the regulation of cellular immune responses against tumors, but it provides the framework to understand the work to be discussed.
Vaccines
Melanoma vaccines have been investigated as adjuvant therapy in resected patients with high risk of recurrence as well as in the therapy for metastatic disease. Generically, vaccines consist of tumor antigen(s) in a formulation designed to activate the cellular arm of the immune system. A common approach has been the use of peptide vaccines of variable length containing the sequence of the target TAAs. These peptides are typically administered with an adjuvant, such as incomplete Freund’s adjuvant or bacterial derivatives like bacillus Calmette-Guérin, which stimulate immune responses nonspecifically. Vaccination strategies using viruses or naked DNA encoding TAAs have been investigated. A spectrum of strategies with DC vaccines have been explored. Irradiated autologous or allogeneic tumor preparations have been used as vaccines. Vaccines have been administered in conjunction with immunostimulatory cytokines such as IL-2, IL-12, and granulocyte-macrophage colony-stimulating factor (GM-CSF).
There are several conceptually appealing features of vaccine strategies: vaccines are most often administered in the outpatient setting, with minimal need for specialized care; they are typically associated with minimal side effects; and, if successful, they may lead to lasting tumor immunity. Investigations in this area have been disappointing in their failure to consistently and clearly show clinical efficacy. Klebanoff and colleagues recently examined notable contemporary cancer vaccine trials reported since 2004; overall, only 12 of 309 (3.9%) patients with melanoma had objective responses. In retrospect, this entire body of work highlights our incomplete understanding of the mechanisms required to trigger antitumor immunity. Through these early failures, there have been substantial gains in fundamental knowledge and promise for a path to improving on immunization strategies.
The lack of efficacy in vaccine trials has been attributed to a variety of shortcomings. First, the central role of DCs in successful immunization was not recognized as cancer vaccines were first being developed, and this point has only recently been addressed in newer vaccine studies. It has been recognized that adjuvant agents are crucial for the initiation of immune responses, yet the array of approved adjuvants remains woefully limited and their function poorly understood. Despite the identification of hundreds of TAAs, it remains unclear which are the best targets to reach the end point of clinical efficacy. Also, antigen expression within any given tumor is heterogeneous and the ability of the tumor to show these antigens is often compromised. Another previously underappreciated component of vaccine strategies is the need to overcome mechanisms of immunosuppression mediated by the tumor or present in the tumor microenvironment. Each successive generation of studies of cancer vaccines has incorporated strategies based in the continuously expanding knowledge of immunobiology.
We have only recently reached the important benchmark of a positive phase 3 study of melanoma vaccines, with 2 promising unrelated studies soon to be reported. Schwartzentruber and colleagues recently published the findings of a multicenter, phase 3 study evaluating the combination of the peptide vaccine gp100 with high-dose IL-2 compared with IL-2 alone. The addition of the peptide vaccine significantly improved response rates (16% vs 6%) and progression-free survival (PFS) (2.2 months vs 1.6 months). There was a trend toward improved median survival (17.8 months vs 11.1 months; P = .06). This monumental effort validates the potential of vaccine strategies in the treatment of established metastatic melanoma but also underscores the need for innovation to enhance efficacy.
Allovectin-7 is a novel vaccine for metastatic melanoma that has shown promise in early-phase clinical studies. Downregulation or loss of expression of MHC class I and β 2 microglobulin molecules has been identified as 1 mechanism through which tumor cells may escape immune detection. Allovectin-7 is a plasmid DNA encoding HLA-B7 and β 2 microglobulin. This vaccine is injected directly into tumors and results in the assembly of a complete MHC class I complex on the tumor surface. It is proposed that the vaccine may have activity through several mechanisms, including expression of a foreign antigen (in HLA-B7–negative patients), increased TAA presentation in the context of HLA-B7, and increased surface expression of MHC class I molecules on the tumor surface (because of the increased availability of β 2 microglobulin molecules). In addition, it is possible that unmethylated bacterial CpG motifs in the plasmid backbone may stimulate immune responses, as has been shown in animal models. Objective response rates of 4% to 11% have been reported in phase 2 studies, with some patients undergoing CR. Antitumor responses have been reproducible in multiple phase 2 trials, and all studies document an extremely favorable toxicity profile. A phase 3 clinical trial comparing Allovectin-7 with DTIC or temozolomide has recently completed accrual.
Another promising vaccine is OncoVEX GM-CSF , an oncolytic herpes simplex virus type 1 that has been genetically engineered for tumor-selective replication and enhanced tumoricidal activity. In addition, the virus encodes the immunomodulatory cytokine GM-CSF, which stimulates the proliferation, maturation, and differentiation of DCs. The vaccine is injected intratumorally. Responses are seen both in the injected lesions and at distant sites. It is presumed that tumor lysis at the site of injection releases antigens, which are subsequently presented by DCs, initiating systemic antitumor effects. In a phase 2 study, the objective response rate was 26%, with more than half of patients achieving CR. Subsequently, an eagerly anticipated phase 3 study has completed accrual, with results expected shortly.
Adoptive Cell Transfer Therapy
Adoptive cell transfer therapy (ACT) for patients with unresectable melanoma has evolved through elegant basic, translational, and clinical research. ACT involves resection of a melanoma metastasis, isolation of tumor-infiltrating lymphocytes (TILs) from the tumor explant, expansion of the T-cell cultures in vitro, and reinfusion of autologous tumor reactive cells together with IL-2 into the patient, after a lymphodepleting preparation. Transgenic mouse models of ACT have played a major role in the optimization of human protocols. Murine studies have clarified desirable characteristics of transferred T cells, the importance of lymphodepletion, and the role of APCs. The Surgery Branch of the National Cancer Institute recently reported their extensive experience with ACT in the therapy for 93 heavily pretreated patients with metastatic melanoma. Objective response rates greater than 70% were achieved in the most recent series. Durable, complete tumor regression was achieved in 22% of patients, with an actuarial 5-year survival of 93% in patients with CR. No other therapy has matched these outcomes.
More recently, genetically modified peripheral blood lymphocytes have been used in ACT studies. Antitumor activity has been reported in patients with melanoma treated with T cells retrovirally transduced with high-affinity T-cell receptors (TCRs) recognizing tumor antigens such as MART-1, NY-ESO-1, and MAGE-A3. Chimeric antigen receptors (CARs) comprise the tumor antigen-binding domain of an antibody molecule fused to the intracellular signaling domain of a TCR. CARs specific for high-molecular-weight melanoma-associated antigen and ganglioside GD3 have shown promise in preclinical testing. CARs have several theoretic advantages over TCR-based gene transfer. Effector cell engagement does not depend on peptide antigen expression and MHC-restricted presentation by the tumor, potential mechanisms by which tumors may escape immune detection. Because they function in an MHC-independent manner, there may be wider applicability to patients as an off-the-shelf reagent. In addition, there is the ability to engineer these CARs to express the intracellular signaling domains of a variety of costimulatory molecules to improve T-cell function and survival.
Immunomodulation
Clinical trials of IL-2 showed that the immune system could be activated nonspecifically to promote tumor regression in patients with melanoma. One implication of these trials is that patients with melanoma frequently have endogenous lymphocytes capable of recognizing tumor antigens; it has also been established that lymphocytes that recognize the tumor frequently reside near the tumor. Substantial evidence indicates that the tumor interacts with responding lymphocytes to subvert the immune response and permit tumor growth, a process referred to as immunoediting.
According to the classic 2-signal model of T-cell activation, the first signal is provided through the interaction of the antigen-MHC complex with the TCR. Naive T cells encountering an antigen must also receive a second signal to initiate an immune response. This process occurs through the interaction of CD28 receptors on T cells with B7-1 (CD80) and B7-2 (CD86) molecules, which are present on APCs. In the absence of costimulation, T cells become anergic. Subsequently, additional costimulatory pathways providing both activating (CD28, OX40, GITR, CD137, CD27, HVEM) and inhibiting (CTLA-4, PD-1, TIM-3, BTLA, VISTA, LAG-3) second signals have been identified. The interplay of these costimulatory molecules serves to precisely control the intensity and duration of cellular immune responses. Elucidation of these diverse pathways has broadened the concept of costimulation, and these immunologic checkpoints represent new targets for therapeutic intervention.
Cytotoxic T Lymphocyte Antigen 4 Blockade
Cytotoxic T lymphocyte antigen 4 (CTLA-4) was identified as a homologue of CD28, which receives the second signal in T-cell activation. In contrast to CD28, it is not present on the surface of naive T cells but is rapidly upregulated with T-cell activation. CTLA-4 binds B7-1 and B7-2 with significantly higher affinity than CD28, and signaling through CTLA-4 engagement serves to attenuate T-cell activation and proliferation. Its native function has been described a brake that prevents productive immune responses from progressing to autoimmune injury; CTLA-4–deficient mice die at 3 to 4 weeks of age from a fatal lymphoproliferative disorder. CTLA-4 is overexpressed on chronically stimulated and exhausted T cells. In addition, CTLA-4 is constitutively expressed on T-regulatory cells, which may be crucial for their suppressive function. Blockade of CTLA-4 results in enhanced T-cell–mediated immune responses, and numerous animal models have shown that tumor immunity can be induced by interfering with CTLA-4 signaling. Subsequently, ipilimumab, a fully human monoclonal antibody, was developed as a therapeutic agent to block CTLA-4. Early-phase clinical trials reported activity in patients with metastatic melanoma.
Ipilimumab received FDA approval in 2011 after the publication of a phase 3 study in which patients with metastatic melanoma were treated with ipilimumab alone or in combination with the gp100 peptide vaccine. The control arm received only the gp100 vaccine. All patients had received previous therapy with other agents, including more than 20% of patients receiving IL-2. There were significant differences in survival with 1-year and 2-year overall survival of 45.6% and 23.5% in patients receiving ipilimumab compared with 25.3% and 13.7% in patients receiving gp100 alone. Overall survival was independent of previous IL-2 therapy, suggesting that these immunomodulating agents act through different mechanisms. Grade 3 or 4 toxicities were frequent (10% to 15%), and death attributed to immune-related adverse events occurred in approximately 1% of patients. The gp100 vaccine did not enhance the efficacy of CTLA-4 blockade, as was seen when this vaccine was combined with IL-2. This finding reinforces the concept that IL-2 and anti-CTLA-4 have inherently different mechanisms of action. This landmark study is the first randomized clinical trial showing a benefit in overall survival for patients metastatic with melanoma. Another pivotal phase 3 study examined the efficacy and safety of ipilimumab plus DTIC compared with DTIC alone in 502 patients with previously untreated metastatic melanoma. The 1-year, 2-year, and 3-year survival were 47.3%, 28.5%, and 20.8% for patients receiving ipilimumab and DTIC compared with 36.3%, 17.9%, and 12.2% for patients receiving DTIC alone. Grade 3 or higher immune-mediated adverse events were documented in 38.1% of patients receiving ipilimumab; there were no deaths attributed to autoimmune phenomena.
Prieto and colleagues recently reported the longest follow-up on patients with melanoma treated with ipilimumab in 3 consecutive trials conducted at the National Cancer Institute. With median follow-up of 71 to 92 months, this study reveals unique and important features of this therapy that are often not apparent in the randomized studies. First, objective responses frequently evolved slowly; in complete responders, the median time to CR was 30 months, with 1 patient achieving CR at 71 months. Reports with shorter follow-up may underestimate the true objective response rate. Second, CRs are remarkably durable, with only 1 of 15 patients recurring after CR; all of the other patients with CR were alive without evidence of disease with median follow-up of 83 months. This finding is reminiscent of the experience with IL-2, and it is tempting to speculate that some of these patients may be cured.
Programmed Death 1/Programmed Death Ligand 1 Blockade
Programmed death 1 (PD-1) and its ligands, PD-L1 and PD-L2, have recently attracted attention as another immunologic checkpoint that can be manipulated to achieve antitumor immune responses. PD-1 was first identified in 1992 and its ligands nearly 10 years later. Signaling through the PD-1/PD-L1(-L2) axis serves to dampen T-cell–mediated immune responses and plays a critical role in central tolerance induction. Inflammation and cardiomyopathy have been observed in PD-1 knockout mice. This autoimmune phenotype is considerably milder than that seen in CTLA-4 knockout mice and does not manifest until 6 to 9 months after birth. PD-1 is not expressed on naive T cells but is induced quickly on activation. When antigen-specific T cells are chronically stimulated, PD-1 is upregulated, and the cells become functionally impaired; T-cell function can be restored with PD-1 blockade. In TILs derived from patients with melanoma, increased PD-1 expression has been correlated with an exhausted phenotype and impaired effector function. PD-L1 is constitutively expressed in hematopoietic cells and a wide variety of other tissues. Because of this distribution, it has been postulated that this pathway serves to protect normal peripheral tissues from activated T cells, thus limiting autoimmunity. PD-L1 is also expressed by many tumors, including melanoma. In solid tumors, PD-L1 can inhibit the cytolytic activity of tumor-infiltrating T cells. Furthermore, PD-L1 expression has been correlated with an unfavorable prognosis in a variety of cancer histologies. Expression of PD-L1 is believed to be a mechanism by which tumors may escape immune rejection.
Two concurrently published phase 1 clinical trials using monoclonal antibodies to disrupt the PD-1/PD-L1 axis have shown promising clinical efficacy across an array of histologies including non–small cell lung cancer, melanoma, and renal cell cancer. In the study reported by Topalian and colleagues, anti-PD-1 antibody was administered in escalating doses to 296 patients. These individuals included 94 patients with melanoma, for whom the objective response rate was 28%. Most responding patients had durable responses of greater than 1 year. Pretreatment tumor specimens were available from a few study patients, and it was noted that objective responses were observed only in patients whose tumors expressed PD-L1, suggesting that this may be a potential biomarker for patient selection. Grade 3 or higher toxicities occurred in 14% of patients. Potential autoimmune toxicities included pneumonitis, vitiligo, colitis, hepatitis, hypophysitis, and thyroiditis. There were 3 cases of fatal pneumonitis. Brahmer and colleagues reported the companion trial, in which patients with a variety of histologies were treated with escalating does of anti-PD-L1 antibody. In the patients with melanoma, 9 of 52 patients had objective responses (17%), including 3 CRs. As reported with the anti-PD-1 antibody, responses tended to be durable, and stable disease (27%) was a frequent outcome in patients not classified as having an objective response. Grade 3 or 4 toxicities occurred in only 9% of study patients. Potentially immune-related toxicities included rash, hypothyroidism, hepatitis, sarcoidosis, endophthalmitis, diabetes mellitus, and myasthenia gravis. Taken together, these studies show that blockade of PD-1 or PD-L1 can result in tumor regression, with a pattern of response similar to that seen with CTLA-4 blockade. The immune-related toxicities attributed to anti-PD-1 and anti-PD-L1 are less infrequent and milder than those associated with anti-CTLA-4. Phase 2 and 3 trials will further define the efficacy and applicability of these therapies, and FDA approval of 1 or both agents seems imminent based on the dramatic results already reported.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


