Iron overload is an inevitable consequence of blood transfusions and is often accompanied by increased iron absorption from the gut. Chelation therapy is necessary to prevent the consequences of hemosiderosis. Three chelators, deferoxamine, deferiprone, and deferasirox, are presently available and a fourth is undergoing clinical trials. The efficacy of all 3 available chelators has been demonstrated. Also, many studies have shown the efficacy of the combination of deferoxamine plus deferiprone as an intensive treatment of severe iron overload. Alternating chelators can reduce adverse effects and improve compliance. Adherence to therapy is crucial for good results.
Key points
- •
Thalassemia major is caused by defects in the synthesis of one or more of the globin subunits of hemoglobin, resulting in variable phenotypes.
- •
The yearly incidence of symptomatic individuals is estimated at 1 in 100,000 people throughout the world (22,989 new births) and 1 in 10,000 people in the European Union.
- •
Patients with thalassemia, being transfusion-dependent and having a hyperactive marrow, accumulate iron in tissues.
- •
The worldwide birth rate of individuals with symptomatic sickle cell disease (SCD) is approximately 2.2 per 1000 births. However, the disease incidence varies between ethnic groups.
- •
Blood transfusions may be required in both acute and chronic complications of SCD.
- •
SCD and thalassemia major differ in iron-loading patterns and in the prevalence of iron-induced organ damage.
Introduction
The natural history of both thalassemia major (TM) and sickle cell disease (SCD) has been completely transformed in industrialized countries by the introduction of modern blood transfusions (filtered red cell concentrates that carry an extremely low residual risk of pathogen transmission) and iron chelation. The life expectancy in TM has changed from a few years to 5 or 6 decades and it may soon equal that of the nonthalassemic population. Mortality continues to decrease, mainly because of a reduction in cardiac deaths ( Fig. 1 ).
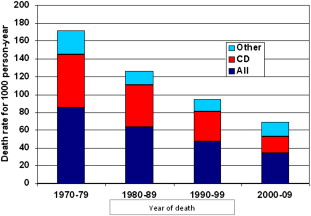
Despite the difficulties they still encounter, patients with SCD have also experienced great improvement in quality of life thanks to the introduction of transfusions and hydroxyurea for the prevention and treatment of several complications.
Blood transfusions
TM
The anemia of TM becomes symptomatic between 6 months and 2 years of age, requiring the institution of a regular transfusion program. As a consequence of continuous anemia, erythropoiesis, although inefficient, can be intense; the bone marrow undergoes an enormous expansion with consequent distortion of facial features, and the plasma volume increases. In addition, hepatosplenomegaly develops. A regimen maintaining a minimum hemoglobin concentration of 9.5 to 10.5 g/dL prevents all the above complications and fosters normal growth at least until puberty.
SCD
Red blood cell transfusions are a mainstay in the treatment of both acute and chronic SCD complications. Most patients receive at least one transfusion in their lifetime, usually for acute complications. Blood transfusions increase arterial oxygen pressure and hemoglobin oxygen-affinity, thereby reducing red cell sickling, and they also improve microvascular perfusion. Regular blood transfusion regimens additionally suppress endogenous erythropoiesis and therefore the production of red cells containing sickle hemoglobin.
Common indications for both acute and chronic transfusions are shown in Box 1 .
Indications for acute transfusions
- •
After surgery and/or general anesthesia
- •
Acute splenic or hepatic sequestration
- •
Aplastic crisis
- •
Acute chest syndrome a,
- •
Stroke or acute neurologic deficit a,
- •
Multiorgan failure a,
- •
Indications for regular transfusion programs
- •
Primary and secondary stroke prevention
- •
Recurrent acute chest syndrome
- •
Progressive organ failure
- •
a May require exchange transfusion.
Erythrocytapheresis in SCD
In SCD, erythrocytapheresis or manual exchange transfusions are alternatives to long-term simple transfusions. Automated erythrocytapheresis is the most accurate method for achieving a target hemoglobin S, but it is also expensive, invasive, and not available in all centers. Manual partial exchange transfusion can be used as an alternative. Both methods slow or prevent further accumulation of transfusional iron.
Introduction
The natural history of both thalassemia major (TM) and sickle cell disease (SCD) has been completely transformed in industrialized countries by the introduction of modern blood transfusions (filtered red cell concentrates that carry an extremely low residual risk of pathogen transmission) and iron chelation. The life expectancy in TM has changed from a few years to 5 or 6 decades and it may soon equal that of the nonthalassemic population. Mortality continues to decrease, mainly because of a reduction in cardiac deaths ( Fig. 1 ).
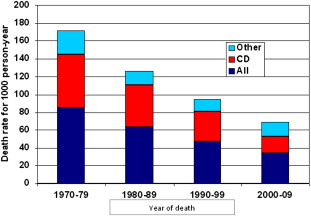
Despite the difficulties they still encounter, patients with SCD have also experienced great improvement in quality of life thanks to the introduction of transfusions and hydroxyurea for the prevention and treatment of several complications.
Blood transfusions
TM
The anemia of TM becomes symptomatic between 6 months and 2 years of age, requiring the institution of a regular transfusion program. As a consequence of continuous anemia, erythropoiesis, although inefficient, can be intense; the bone marrow undergoes an enormous expansion with consequent distortion of facial features, and the plasma volume increases. In addition, hepatosplenomegaly develops. A regimen maintaining a minimum hemoglobin concentration of 9.5 to 10.5 g/dL prevents all the above complications and fosters normal growth at least until puberty.
SCD
Red blood cell transfusions are a mainstay in the treatment of both acute and chronic SCD complications. Most patients receive at least one transfusion in their lifetime, usually for acute complications. Blood transfusions increase arterial oxygen pressure and hemoglobin oxygen-affinity, thereby reducing red cell sickling, and they also improve microvascular perfusion. Regular blood transfusion regimens additionally suppress endogenous erythropoiesis and therefore the production of red cells containing sickle hemoglobin.
Common indications for both acute and chronic transfusions are shown in Box 1 .
Indications for acute transfusions
- •
After surgery and/or general anesthesia
- •
Acute splenic or hepatic sequestration
- •
Aplastic crisis
- •
Acute chest syndrome a,
- •
Stroke or acute neurologic deficit a,
- •
Multiorgan failure a,
- •
Indications for regular transfusion programs
- •
Primary and secondary stroke prevention
- •
Recurrent acute chest syndrome
- •
Progressive organ failure
- •
a May require exchange transfusion.
Erythrocytapheresis in SCD
In SCD, erythrocytapheresis or manual exchange transfusions are alternatives to long-term simple transfusions. Automated erythrocytapheresis is the most accurate method for achieving a target hemoglobin S, but it is also expensive, invasive, and not available in all centers. Manual partial exchange transfusion can be used as an alternative. Both methods slow or prevent further accumulation of transfusional iron.
Iron overload
Transfusional iron intake amounts to approximately 0.3 to 0.5 mg/kg/d for most patients. The transfusion iron burden can be calculated as the total amount of pure red blood cells transfused (volume of packed red cells multiplied by the hematocrit [%] of the unit) × 1.08. Most units of transfused red cells contain 180 to 200 mg of iron.
Part of the non-transferrin-bound iron circulating in the plasma (labile plasma iron) produces reactive radicals, and the resulting oxidation products are thought to be responsible for a large part of the iron accumulation and parenchymal cell injury associated with regular transfusions. The organs particularly affected are the heart, liver, pancreas, and endocrine glands.
Based on the experience collected in hereditary hemochromatosis, a liver iron concentration (LIC) of 7 mg/g was considered compatible with a normal life expectancy and therefore an appropriate target for control of iron overload with chelation therapy. Today, however, on the basis of numerous studies of magnetic resonance imaging (MRI), it is clear that the LIC does not consistently reflect the cardiac iron content and therefore cannot accurately predict prognosis. Without effective iron chelation, death commonly occurs in the second or third decade in patients with TM, usually because of iron-induced cardiac disease.
Does Iron Overload Differ Between SCD and TM?
Iron metabolism, and consequently, patterns of iron loading differ in SCD and TM. Erythropoiesis is variably increased in SCD, but it is not ineffective. Unlike patients with thalassemia, patients with nontransfused SCD do not develop systemic iron overload because of increased iron absorption, and they may actually present iron deficiency, possibly related to intravascular hemolysis and the resulting excessive urinary loss of iron.
Inflammation, which is part of the pathophysiology of SCD, increases synthesis of hepcidin and consequently decreases iron absorption and enhances retention of iron within the reticulo-endothelial system. As a result, tissues and organs affected by iron overload are different in SCD and TM. Specifically, iron-induced cardiac and endocrine dysfunction is less common in SCD.
Transfusion regimens may sometimes differ in the 2 populations. Patients with TM may require transfusions every 2 to 4 weeks to maintain hemoglobin values between 9 and 10 g/dL, beginning at a very young age (3–6 months). On the other hand, in SCD regular transfusions are usually started later in life, and many patients, particularly adults, receive occasional simple transfusions rather than transfusions on a regular schedule.
Iron-induced Organ Damage in SCD
As noted above, organ damage secondary to iron overload has been found to be disease-specific. In a retrospective study, Vichinsky and colleagues reported a higher prevalence of cardiac disease (20% vs 0%), growth failure (27% vs 9%), and endocrine failure (37% vs 0%) in 30 patients with TM compared with 43 patients with SCD. In a multivariate analysis, the only significant predictors of combined endocrine and cardiac disease were duration of chronic transfusion and diagnosis.
Oxidative stress also differs in the 2 diseases. A study investigated the relationship between iron overload and disease-specific organ damage in TM and SCD by analyzing biomarkers of oxidative stress, inflammation, and tissue injury. Inflammation in SCD induced higher levels of the anti-oxidant γ-tocopherol compared with TM, leading to decreased tissue peroxidation and injury.
Complications of Iron Overload
Heart
Cardiac problems are a frequent consequence of iron overload and in particular of the peroxidative damage to lysosomes and mitochondria by the labile iron pool. A recent Italian cooperative study demonstrated that 22% of patients with TM undergoing MRI had one or more cardiac problems, including heart dysfunction (66%), arrhythmias (14%), and both heart dysfunction and arrhythmias (19%). The prevalence of cardiac disease was significantly higher in male than in female patients ( Fig. 2 ) and was dependent on the cohort of birth. Heart failure and arrhythmias have been responsible for more than 70% of all deaths ( Fig. 3 ). An expert consensus from the American Heart Association on the diagnosis and treatment of cardiac dysfunction in TM has been recently issued.
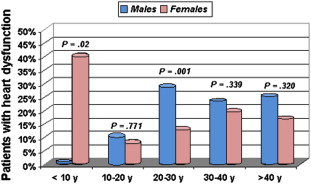
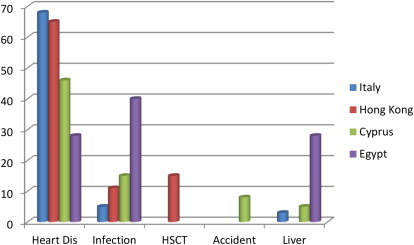
Increasing literature suggests that patients with SCD may be protected from iron-related cardiac damage. Several MRI studies demonstrated an absence of cardiac iron in patients with SCD. Wood and colleagues found that heavily iron-loaded patients with SCD had normal cardiac T2* values despite monthly transfusions for an average of 9.3 years. More recently, similar findings were described in 23 Lebanese patients with SCD (mean cardiac T2* = 37.3 ± 6.2 ms; range: 21.9–46.8 ms), even in the presence of significant systemic and hepatic iron overload. However, in regularly transfused patients with SCD, effective chelation therapy is still needed to avoid hepatic iron overload and iron-related organ dysfunction.
Liver
Liver iron overload is a cause of fibrosis and cirrhosis, especially when associated with blood-transmitted hepatotropic viruses, in particular HCV. Cirrhosis is a major risk factor for liver failure and can evolve to hepatocellular carcinoma. The prevalence of cirrhosis in patients with TM ranges from 10% to 20%. However, as described earlier, the LIC does not always correlate well with cardiac iron and should therefore not be used alone to predict risk for iron-related cardiac disease.
The hepatic pattern of iron distribution differs in patients with SCD compared with patients with TM. In a recent histologic study, patients with SCD were found to have significantly less hepatic fibrosis and inflammation at similar levels of LIC as patients with TM. Although patients with SCD had predominantly sinusoidal iron deposition, parenchymal iron deposition was observed even at low LIC. Iron-related hepatic damage in patients with SCD may also be enhanced by the high incidence of transfusion-acquired hepatitis C in some developing countries.
Endocrine organs
In TM, iron deposition and structural damage of the pancreas the pituitary, parathyroid, thyroid, and adrenal glands and the gonads have been demonstrated histologically and by MRI. The reported prevalence of endocrine complications in TM is shown in Fig. 4 .
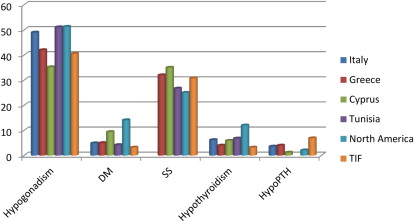
Despite iron overload, endocrinopathies are less frequent in SCD. A study comparing iron overloaded subjects with TM and SCD confirmed a higher prevalence of endocrinopathies in patients with TM. Moreover, the rate of endocrine disorders was similar in transfused and nontransfused patients with SCD, suggesting a role of the disease in modulating iron-related endocrine injury.
Iron chelation
To prevent hemosiderosis, iron must be chelated and excreted. Successful long-term iron chelation therapy depends on the achievement of neutral or negative iron balance, meaning the amount of iron excreted with chelation therapy must be the same or more than the amount of iron acquired from transfusion and gastrointestinal absorption. Because the mean iron intake differs even among patients with the same hematologic disorder because of factors such as molecular defect, presence of red cell alloantibodies, spleen size, and other factors, it is important to know the actual transfusional iron intake for a patient when choosing the dose of chelator. Three iron chelators are now commercially available ( Tables 1 and 2 ) and a fourth is undergoing phase 3 trials ( Box 2 ).
| Deferoxamine | Deferiprone | Deferasirox | |
|---|---|---|---|
| US Food and Drug Administration (FDA) approval | 1982 (500 mg vial); 2000 (2 g vial) | 2011 | 2005 |
| European Medicines Agency (EMEA) approval | — | 1999 | 2006 |
| Brands and trademarks | Desferal, Novartis Generic products by several manufacturers (Bedford, Fresnius Kabi USA, Hospira, Watson Labs) | Ferriprox, Apotex Inc Kelfer, Cipla Ltd, India | Exjade, Novartis |
| Molecular structure | N-[5-[3-[(5-aminopentyl) hydroxycarbamoyl] propionamido]pentyl]-3[[5-(N-hydroxyacetamido)pentyl]carbamoyl] propionohydroxamic acid monomethanesulfonate | 3-hydroxy-1,2-dimethylpyridin-4-one | 4-[3,5-Bis(2-hydroxyphenyl)-1H-1,2,4-triazol-1-yl] benzoic acid |
| Indications | Acute iron intoxication Chronic iron overload secondary to transfusion-dependent anemias | Chronic iron overload in patients with TM when deferoxamine (DFO) is contraindicated or inadequate |
|
| Age | None | Limited data <6 y of age | >2 y of age |
| Dose | 20–50 mg/kg/d SC over 8–24 h through a portable pump | 75–99 mg/kg/d in 3 daily doses | 10–40 mg/kg/d in single dose, according to transfusional iron intake |
| Available formulations | 500 mg and 2 g vials of DFO mesylate USP in sterile, lyophilized form | 500 mg and 1 g film-coated tablets 100 mg/mL oral solution | 125 mg, 250 mg, 500 mg tablets for oral suspension |
| Metabolism | Plasma enzymes | Liver glucuronidation | Uridil glucuronosyltransferase enzymes |
| Iron excretion | Mainly urine; in some patients fecal excretion up to 40% | Mainly urine | Mainly feces |
| Drug | Adverse Effects | Management | Recommendations |
|---|---|---|---|
| DFO | Local side effects at the subcutaneous injection site | Verify correct positioning of needle; hydrocortisone infused with DFO | Adjust dose to serum ferritin levels Monitor weight, height, and growth velocity every 3 mo in children Visual acuity tests, slit-lamp examination, and fundoscopy annually Audiometry annually Monitor renal function periodically |
| Stunted growth and bone changes (eg, metaphyseal dysplasia) | Reduce dose or change chelator | ||
| Hypersensitivity reactions and systemic allergic reactions | Change treatment drug or desensitization | ||
| High-frequency sensorineural hearing loss | Rare if dosage guidelines followed and dose adjusted when ferritin declines; if abnormal tests or symptoms, discontinue treatment | ||
| Visual disturbance | |||
| Increased serum creatinine, acute renal failure, and renal tubular disorders | Reduce dose or discontinue treatment | ||
| Increased susceptibility to Yersinia enterocolitica and pseudotuberculosis and Klebsiella pneumoniae infections | Discontinue treatment if suspected signs and symptoms. Initiate antibiotic therapy | ||
| DFP | Nausea, abdominal pain, vomiting, diarrhea | Usually transient without need to modify treatment; otherwise reduce dose and then increase gradually; as an alternative, switch to oral solution | Weekly neutrophil count Monitor hepatic and renal functions periodically Monitor weight and body mass index Avoid use with other neutropenia-inducing drugs |
| Neutropenia (neutrophils 0.5–1.5 × 10 9 /L) | Discontinue DFP and repeat neutrophil count daily until normalization; rechallenge with caution | ||
| Agranulocytosis (neutrophils <0.5 × 10 9 /L on 2 consecutive tests) | Immediately discontinue DFP; if signs/symptoms of infection, perform cultures and begin antibiotic treatment; granulocyte colony stimulating factor may be indicated | ||
| Increased liver enzymes | Usually asymptomatic and transient without need to modify treatment but may require lowering dose and gradually raising again | ||
| Increased appetite | Dietary measures | ||
| Zinc deficiency | Oral zinc supplementation | ||
| Arthralgia and arthropathy | Mild arthralgia is usually transient; if more severe disease, reduce dose or discontinue treatment | ||
| Neurologic disorders | Described in children taking long-term 2.5 times maximum recommended dose, often in absence of systemic iron overload; discontinue DFP | ||
| DFX | Nausea, vomiting, and abdominal pain | Usually transient; if needed reduce dose or discontinue treatment | Monitor serum creatinine, creatinine clearance, and/or plasma cystatin C twice before initiation, weekly in the first month after initiation, or modification of therapy and then monthly Monthly urinalysis for proteinuria monthly Monitor renal tubular function as needed Monitor serum transaminases, bilirubin, and alkaline phosphatase before initiation of treatment, every 2 wk during the first month, and then monthly Auditory and ophthalmic testing annually |
| Diarrhea (may be due to coexistent lactose intolerance) | Consider administration with lactase-containing products | ||
| Rash | Discontinue DFX; reintroduce at a lower dose after resolution (if necessary add short course of corticosteroids) | ||
| Increased serum creatinine, proteinuria, acute renal failure, renal tubulopathy, Fanconi syndrome | Reduce dose by 10 mg/kg if increase in serum creatinine by >33% above pretreatment average and if estimated creatinine clearance decreases below the lower limit of normal at 2 consecutive visits or if abnormalities of tubular markers; if no improvement, change chelator. For more severe changes, consider discontinuing DFX | ||
| Increased liver enzymes, hepatic failure | If persistent and progressive increase in serum transaminases, interrupt treatment; once normalized, cautious reinitiation at a lower dose | ||
| Upper gastrointestinal ulceration and hemorrhage | Observe carefully in patients in treatment with other potentially ulcerogenic drugs; discontinue treatment if signs or symptoms | ||
| Auditory (decreased hearing) and ocular (lens opacities) disturbances | Reduce dose or discontinue treatment | ||
| Cytopenias | Discontinue treatment |
- •
Severity of iron overload
- •
Transfusional iron intake
- •
Effectiveness
- •
Toxicity
- •
Chelator availability
- •
Route of administration
- •
Adherence
- •
Patient’s preference
- •
Cost
Deferoxamine B
DFO, a hydroxamic acid and hexadentate chelator, was developed over 50 years ago as the first available chelating agent ( Box 3 ). It did not undergo the formal trials that would currently be required before being approved by the regulatory agencies. The extraordinary effect of the introduction of chelation in clinical practice is demonstrated by the change in survival of patients with TM subdivided by cohort of birth. In fact, a very marked improvement was seen for patients born after 1974 who had the benefit of subcutaneous chelation from the first years of life. Treatment with continuous intravenous DFO has been shown to improve myocardial iron, even in the most overloaded hearts with average myocardial T2* values of less than 6 ms, and to rescue patients in congestive heart failure. The main limit of DFO is the inconvenience of parenteral administration. The patients’ compliance, therefore, is often erratic, especially during the teenage years.
- •
DFO was the first chelating agent introduced in clinical practice.
- •
It has a large molecular weight and a short plasma half-life.
- •
It is not efficiently absorbed from the gut and negative balance is rarely obtained after intramuscular injection; therefore, it is now usually administered subcutaneously, by means of a portable, battery-operated pump or an elastomeric balloon, over 8–12 hours at night.
- •
DFO as a continuous intravenous infusion is very effective in reversing severe cardiac iron overload and iron-induced congestive heart failure.
- •
It efficiently binds non-transferrin-bound iron, preventing free radical formation and lipid peroxidation.
- •
In patients with SCD, the safety and efficacy of high-dose intravenous DFO were demonstrated by liver biopsy.
Related posts:




Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


