Antigen
Antigen class
Phase
Comment
Alpha fetoprotein (αFP)
Differentiation
Preclinical
Carcinoembryonic antigen (CEA)
Differentiation
Ph1
Terminated—poor accrual
Glycoprotein 100 (Gp100)
Differentiation
Ph2
Hepatitis B virus (HBV) antigen
Viral antigen
Ph1
Human papilloma virus (HPV) early protein 6 (E6)
Viral antigen
Ph1
HPV E7
Viral antigen
Ph1
Melanoma-associated antigen (MAGE) A3
CGA
Ph1
TCR assets in development cover multiple HLA types: HLA-A*01, A*02, A*24, and DP4.
Different TCRs have varying degrees of cross-reactivity to MAGE A6, A9, and A12.
One candidate caused patient deaths due to cross-reactivity to MAGE A12 in the brain. Another asset caused patient deaths due to cross-reactivity with titin in heart
MAGE A4
CGA
Ph1
TCR assets in development cover two HLA types: HLA-A*02 and A*24
MAGE A10
CGA
Ph1/2
Melanoma antigen recognized by T cells (MART)-1/melan-A
Differentiation
Ph2
New York esophageal antigen-1 (NY-ESO-1)
CGA
Ph1/2
Preferentially expressed antigen in melanoma (PRAME)
CGA
Preclinical
p53
Overexpressed mutant form acts as an oncogene
Ph2
Thyroglobulin
Differentiation
Ph1
Suspended
Tyrosinase
Differentiation
Ph1
Wilms tumor 1 (WT-1)
Oncogene/TAA
Ph1/2
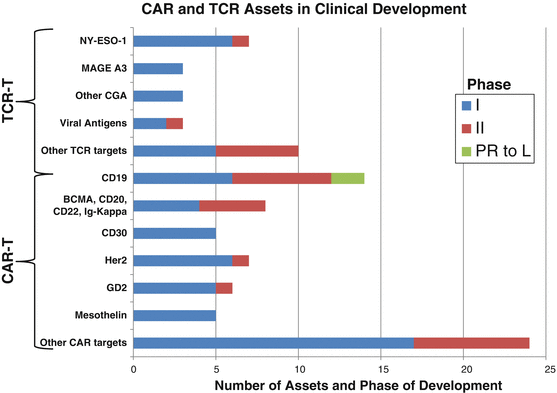
Fig. 42.1
The pipeline of CAR T cell and TCR T cell therapies. Thus far the CAR T cell pipeline is both broader and more mature than the TCR T cell pipeline. Following the success of CD19, other B cell lineage antigens (BCMA, CD20, CD22, and immunoglobulin kappa chain) dominate, and CD30, while strictly a TAA, is being used to target the B cell malignancies Hodgkin’s and non-Hodgkin’s lymphomas. The cancer germline antigens dominate the TCR T cell pipeline. PR to L pre-registration to launch. Sources: clinicaltrials.gov and Pharmaprojects® | Pharma Intelligence, 2017
Looking beyond the classes of antigen that have been exploited for TCR T cell therapies, thus far, there are a couple of potential target classes that seem especially promising: patient-specific neoepitopes, including aberrant tumor-associated posttranslational modifications, and T cell epitopes associated with impaired peptide processing (TEIPP).
With the evolution of deep sequencing, mass spec, and bioinformatic technologies, it is becoming feasible to identify the neoepitopes unique to a given patient’s tumor and to expand autologous T cells recognizing these private tumor mutations from patient blood [42–44]. This may result in an effective method to implement a polyclonal TIL-like approach and/or identify some neoepitopes that are shared with larger groups of patients for a TCR T cell approach. While many neoepitopes result from amino acid substitutions, frameshifts, or alternate/cryptic open reading frames, altered posttranslational modification resulting in phosphopeptides and arginine (di)methylated peptides have been recently identified as promising target classes [42, 45, 46] and should be shared across a greater proportion of patients as they are not reliant on specific mutations but upon altered posttranslational modifications, which frequently occur in tumors. Indeed, aberrant protein phosphorylation is a hallmark of tumor cells and creates numerous phosphopeptide antigens that can be recognized as distinct from their non-phosphorylated counterparts by conventional αβ T cells.
Under selective pressure from TILs or therapeutic TCR T cells, tumor variants that have lost or downregulated antigen presentation by epigenetic or genetic mutations may prevail. While HLA deletion renders tumors refractory to T cell therapy, it may make them more vulnerable to NK cells. Downregulation or loss of antigen processing pathway components other than MHC is frequently observed and may represent a means to escape both T and NK cells. However, in the setting of MHC expression without antigen processing, tumors may present a unique and broadly shared set of target peptides—the TEIPP [47]. So far suitable epitopes have only been identified in mice, where it has been shown that cells defective for the transporter associated with antigen processing (TAP) present low levels of MHC class I molecules complexed with a peptide from the TRH4 protein. In the presence of TAP, the binding of the TRH4 peptide to the MHC class I molecule Db is easily outcompeted by numerous more suitable peptides from the proteome of the cell, but without TAP to transport these peptides into the ER, sufficient TRH4-Db peptide complexes are displayed on the cell surface where they can activate high avidity T cell clones. Such clones exist because they escaped negative thymic selection due to inability of TAP-replete cells to present such peptide-MHC complexes [48].
While the CAR T cell field has pursued a number of the antigens shared with the cancer vaccine field, a key limitation has been that only cell surface targets are tractable for CARs. As described above, lineage-restricted targets have been exploited to target B cell malignancies. There is intense competition around relatively few targets that have attractive efficacy and acceptable safety profiles for B cell malignancies (Fig. 42.1). Beyond these, there are numerous tumor-associated antigens and differentiation antigens that are being explored with more clinical data becoming available in the coming years (summarized in Table 42.2 and Fig. 42.1).
Table 42.2
Antigens targeted thus far by CAR T cells in clinical trials
Antigen | Antigen class | Phase of development | Comment |
|---|---|---|---|
B cell maturation antigen (BCMA) | Differentiation | Ph2 | |
Carbonic anhydrase IX (CAIX) | TAA | Ph1 | |
CEA | Differentiation | Ph1 | |
CD7 | TAA | Ph1/2 | |
CD19 | Lineage-specific antigen | Ph3–launch | |
CD20 | Lineage-specific antigen | Ph2 | |
CD22 | Lineage-specific antigen | Ph1 | |
CD30 | TAA | Ph1/2 | |
CD33 | TAA | Ph1/2 | |
CD70 | TAA | Ph1/2 | |
CD123 | TAA | Ph1 | Expression on pluripotent stem cells poses risks |
CD171 (L1-cam) | TAA | Ph1 | |
c-MET (tyrosine protein kinase MET) | TAA | Ph1 | |
Disialoganglioside (GD)2 | TAA | Ph1 | |
Epidermal growth factor receptor (EFGR) | TAA | Ph1 | |
EGFR variant III (EGFRvIII) | Neoepitope | Ph1/2 | |
Ephrin type-A receptor 2 (EphA2) | TAA | Ph1 | |
Fibroblast activation protein (FAP) | TAA | Ph1 | Expressed by fibroblasts in tumor microenvironment rather than tumor cells |
Folate receptor alpha (FRA) | TAA | Ph1 | |
Human epidermal growth factor receptor 2 (Her2Neu) | TAA | Ph1/2 | One asset terminated due to patient death |
Glypican 3 (GCP3) | TAA | Ph1/2 | |
Interleukin 13 receptor α2 | TAA | Ph1 | |
Kappa immunoglobulin | TAA | Ph1 | |
Mesothelin | TAA | Ph1 | |
Mucin (Muc) 1 | Glycoantigen | Ph1/2 | |
Prostate stem cell antigen (PSCS) | Differentiation | Ph1 | |
Prostate-specific membrane antigen (PSMA) | Differentiation | Ph1 | |
Receptor tyrosine kinase orphan receptor (ROR) 1R | TAA | Ph1 | |
Vascular endothelial growth factor receptor (VEGFR) 2 | TAA | Ph1/2 |
In summary, a couple of suitable targets have already been identified in clinical trials; current nonclinical and clinical work is actively addressing a significantly expanded target space, and there are still areas or untapped potential that come from new and less studies target classes as well as from the potential to utilize mutated neo-antigens in the future. This makes us confident that an increasing number of suitable targets covering multiple cancers will become available over the next years and enable fill and flow of future drug development pipelines. Before we revisit the next two key challenges that limit broad use of engineered T cells in oncology, we would like to highlight the key characteristics of the nonclinical and clinical development of CAR T cell and TCR T cell approaches.
42.4 Nonclinical Development
Preclinical development of gene-engineered CAR- and TCR-modified T cells is truly a “first in class” living medicine. Traditional evaluations applied to pharmacologic small molecule inhibitors or even biologics including vaccines and mAb therapies do not apply. Neither do usual evaluations of cellular therapies such as blood transfusions or bone marrow transplants. A third class of medicines that have fairly recently made an impact include gene therapies, generally used to treat single-gene defects in patients with rare diseases. Immunotherapy with CAR and TCR T cells involves aspects of all of these, brought together for the first time. Biologic pharmaceuticals provide the mAb binding region, while gene engineering borrows the use of transforming vectors such as gamma-retro or lentivirus to combine targeting receptor with functional genes. All of these come together to engineer patient T lymphocytes ex vivo to generate a new “living drug” that relies upon millennia of built-in evolution to combat disease inside the patient, in this case, cancer.
There are several steps in common to developing any preclinical CAR or TCR therapy. First consideration is the target. Engineered T cell targets should be expressed on a reasonable proportion of tumors and should not be expressed on critical normal tissues. This became frankly apparent in the first-in-human treatment of a 39-year-old woman with metastatic melanoma who received a high number (1010) of autologous HER2 CAR T cells [36]. Although to date, over 400,000 women with breast cancer have received trastuzumab anti-HER2 mAb in the adjuvant setting, with minimal side effects, the first patient to receive the same HER2 mAb-redirected CAR T cells succumbed to severe respiratory distress, followed by multiple organ failure starting 15 minutes after infusion [36]. This is a clear demonstration of the difference in impact of a naked antibody compared with an antibody tied to the signaling region of a T cell. While there may be mitigating circumstances, whereby utilizing a limited route of administration [49], lowering the affinity of a CAR receptor may allow for selective destruction of high-antigen expressing tumors while sparing low-antigen bearing normal tissues [50, 51], the safest approach is to target truly tumor-specific antigens.
Several methods have been employed to identify preclinical targets on tumors. Initially, many institutions had to rely on their own available tumor banks for antigen detection by immunohistochemical (IHC) or RT-PCR for gene expression. More recently, however, numerous in silico bioinformatic resources have become widely accessible, in particular evaluation through publicly available Human Protein Atlas (proteinatlas.org) or via The Cancer Genome Atlas (TCGA) (cancergenome.nih.gov).
Upon selecting a target, a binder is required, usually in the form of the scFv of a mAb. Once selected, the binder/scFv is engineered into a DNA construct including a linker, hinge/transmembrane domain (typically derived from CD4, CD8, or an IgG), and followed by a co-stimulatory molecule and CD3ζ at minimum, though new additions are continually being produced. The difference between a CAR and a TCR is that a TCR requires expression of a matching full-length CD3 α and β chain comprising the constant and variable regions of each. Only upon appropriate α/β pairing is antigen recognition conferred, implementing the induction of the full CD3 signaling mechanism.
In vitro, recombinant TCR or CAR are expressed in donor T cells, and upon confirming surface expression, these cells undergo specificity recognition and functional testing. At minimum, known antigen-positive and antigen-negative target cells are needed, though in practice, often several panels of different tumor types are assessed for targeting. While there are many variations of experimental methods to determine function, they fall in one of two forms: (1) effector T cell stimulation or (2) target cell destruction. Once they have passed this initial specificity test, they must be evaluated for safety. Two types of safety predominate, specifically (1) on-target, off-tumor, whereby the antigen being targeted is also expressed on normal tissues, or (2) off-target toxicity, in this case the receptor recognizes something other than the specific target. Each type of safety testing poses its own set of challenges, including the need to profile each cell/tissue type with regard to gene expression. In the case of “on-target, off-tumor,” essentially a screening panel of live normal human cell types and tissues are needed for use as experimental targets for the CAR in question. Some groups have utilized human-derived primary cell lines for this [52]; while others have obtained direct primary cell and tissue grafts from donors for this purpose [20, 53].
Off-target toxicity is more difficult to predict and has been observed more for TCRs than for CARs. Part of the reason for this may be (1) the potential for more than one gene/protein to share sequences that can be presented in the context of MHC in a similar conformation to tumor-derived epitopes and (2) the act of “affinity tuning” TCR toward higher affinity for a specific peptide-MHC complex which may introduce unforeseen cross-reactive binding to peptide-MHC complexes from other proteins expressed in normal tissues that would otherwise have been screened out biologically during thymic selection. One such example of this was observed in the affinity maturation of a TCR to a MAGE-A3 peptide-HLA-A*01 complex [41]. Subsequent analyses suggested that the fatalities were likely resulted from recognition of an off-target peptide from the muscle protein titin, highly expressed in cardiac muscle [34]. New methods to predict cross-reactivity profiles for new TCRs such as the regular use of 3D culture models for toxicity-relevant tissues and systematic scans of permutated peptides and use of in silico predictions to predict cross-reactivity profiles for novel TCRs have now become a standard requirement of preclinical development programs. While normal cell/tissue toxicity detection methods still continue to evolve, the progress in developing new models and safety technologies for this type of predictive evaluation will prevent unforeseen adverse events in patients in the future.
In vivo models have some, albeit limited, benefit for translation of CAR and TCR therapies, in that immune-compromised inbred mouse models such as the NOD-SCID-common gamma chain k/o (NSG) or NOG mouse can engraft both human tumors and human TCR or CAR T cells. These animals act as tiny incubators complete with functioning cardio-vasculature and an extensive network of blood vessels to transport the T cells to the tumors. Generally, however, these models are limited to showing antitumor efficacy and homing to tumor sites, and most recently have shown some promise in modeling T cell exhaustion and checkpoint blockade at the tumor site. Unfortunately, few tumor target epitopes are shared between mouse and human, making it difficult to determine information on safety or potential normal tissue toxicity [54]. Syngeneic mouse models with congenic tumors may provide a unique ability to evaluate both scientific questions about the efficacy of CAR therapy, and the specific mechanism of action of downstream cancer “cures,” in the context of an animal model with an intact immune system and physiologic expression of tumor antigen in both tumors and normal tissues, if present [55–58]. However, these models are far from perfect: (1) they need separate binders that likely have different affinity for target than human counterparts; (2) they require completely different vector constructs encoding murine genes for CD3ζ, 4-1BB, and CD28 in place of their human orthologs; and last but not least, (3) mice are not people. There has been much divergent evolution in the development of their immunity and physiology, so results in a mouse model are by no means a guarantee of what will work for patients. To date the use of nonhuman primates (NHP) has been of minimal/negligible benefit in determining safety or toxicity to this field, primarily due to the lack of similarity of action and reagent comparability between human and NHP and also due to the lack of any NHP tumor models. Intriguingly, according to the US Food and Drug Administration (oncology), they currently do not require any animal modeling to support CAR or TCR first time in human phase I clinical trials.
42.5 Gene-Engineered T Cell Therapy: Clinical Development
42.5.1 Chimeric Antigen Receptor T Cell Therapy
The most basic first-generation CARs that were introduced to clinical trials more than 15 years ago comprised a scFv fused to the intracellular CD3ζ TCR signaling chain via a short transmembrane domain. CAR T cells built that way exhibited cytotoxic activity upon antigen recognition but lacked persistence. CARs currently being tested in clinical trials are second-generation constructs that result from the addition of a co-stimulatory signal to the constructs. The vast majority of current clinical trials are assessing the activity of second-generation CARs bearing either 4-1BB or CD28 motifs. Third-generation CARs, including more than one co-stimulatory domain, and fourth-generation CARs, including additional choices of co-stimulatory domain such as OX40 or CD27, are beginning to be tested in clinical trials, though it is too soon to know whether they will show any real benefits over the second-generation designs.
CD19 is expressed on the surface of most B cell leukemias and lymphomas and has emerged as the most attractive for adoptive T cell strategies as it is expressed in normal B cells but no other normal tissues. A number of academic centers are developing CD19-directed CAR T cell clinical programs. Exciting preliminary results have resulted in multiple academic/pharmaceutical industry partnerships that have facilitated the launching of registration studies (Table 42.3).
Table 42.3
Selection of the most clinically advanced CD19-directed CAR T cell programs
Company | Novartis | Juno | Juno | Kite |
|---|---|---|---|---|
Academic partner | University of Pennsylvania | Memorial Sloan Kettering Cancer Center | Fred Hutchinson and Seattle Children’s Hospital | National Cancer Institute |
CD19-targeted CAR | CTL019 | JCAR015 | JCAR017 | KTE-C19 |
Vector platform | Lentivirus | Retrovirus | Lentivirus | Retrovirus |
Anti-CD19 scFv | FMC63 | SJ25C1 | FMC63 | FMC63 |
Hinge/transmembrane domain | CD8-CD8 | CD28 | IgG4-CD28 | CD28 |
Co-stimulatory motif | 4-1BB | CD28 | 4-1BB or CD28 | CD28 |
T cell source | Autologous | Autologous | Autologous | Autologous |
Suicide capability | None | None | EGFRt | None |
The striking clinical activity of CD19 targeted CAR T-cells containing 4-1BB costimulation (CTL019) was first shown in three patients with heavily pretreated chronic lymphocytic leukemia (CLL) by investigators at the University of Pennsylvania [11]. Long-term follow-up study showed that 8 of 14 patients responded (overall response rate [ORR] 57%), including 4 patients with complete response (CR) with no evidence of minimal residual disease (MRD), and that CAR T cells persisted for years [4]. In this study, no patient in CR has yet relapsed, hinting at the curative potential of this cell therapy approach. More than 45 patients with relapsed or refractory CLL have been treated at the University of Pennsylvania, with an ORR of 45% [59]. Other groups have shown similar activity in CLL in smaller patient cohorts [60–62].
Several groups using different CAR T cell designs have reported very high CR rates in both pediatric and adult patients with relapsed/refractory acute lymphoblastic leukemia (ALL). The Children’s Hospital of Philadelphia and the University of Pennsylvania have reported on 30 patients (25 pediatric and 5 adult) with ALL treated with CTL019 [3]. CR was achieved by 27 (90%) patients, and the probability of survival at 6 months was 78%. Durable remissions up to 24 months were observed and correlated with persistent CAR T cells. The National Cancer Institute treated 45 children and young adults with 19–28z CAR T cells, reporting a CR rate of 60% [63]. At the time of the analysis, all patients remained alive and 89% remained disease-free (range 5–28 months). The Memorial Sloan Kettering Cancer Center group has reported on 45 adult patients with relapsed ALL treated with JCAR015, a CD19-targeted CAR containing the co-stimulatory molecule CD28. A CR was achieved by 82% of patients, although CAR persistence was limited to a few months [64]. At Fred Hutchinson Research Cancer Center, 27 of 29 (93%) patients with relapsed ALL achieved CR after infusion of 4-1BB containing CAR T cells given at a 1:1 ratio of CD8+:CD4+, and 25 of them (86%) had no evidence of MRD [65].
CD19-directed CAR T cell therapy is also active in heavily pretreated patients with non-Hodgkin’s lymphoma (NHL). At the National Cancer Institute, the ORR among nine patients with relapsed/refractory diffuse large B cell lymphoma (DLBCL) was 67% [61], whereas at the University of Pennsylvania, it was 47% among 15 patients with DLBCL and 73% among eight evaluable patients with relapsed/refractory follicular lymphoma [66]. In the latter study, no patient achieving CR has yet relapsed.
Results from the pivotal study ZUMA-1, a multicenter pivotal study sponsored by Kite, testing the CD28 containing CAR T cell KTE-C19 (axicabtagene ciloleucel, Axi-cel), have been reported [67]. A total of 101 patients with aggressive NHL (77 with DLBCL and 24 with primary mediastinal B cell lymphoma or transformed follicular lymphoma) received lymphodepletion with fludarabine and cyclophosphamide followed by Axi-cel at 2 × 106 cells/kg. The ORR was 82%, including a CR rate of 54%, with an OS rate at 6 months of 80%. At the time of data cutoff, 44% of patients remained in remission [67]. The ZUMA-1 findings have been submitted to the FDA to support a biologics license application for Axi-cel for the treatment of transplant-ineligible patients with relapsed or refractory aggressive NHL.
Collectively, these results demonstrate the successful application of CAR T cell therapy in CD19+ B cell malignancies but also raise a red flag regarding potential life-threatening toxicities. Nonetheless, the risk/benefit ratio greatly favors the use of CAR T cells in malignancies such as relapsed/refractory ALL or DLBCL for which effective therapies are desperately needed. Likely, the available clinical trial results with CD19-directed CAR T cells will lead to the first regulatory approval of a gene-engineered adoptively transferred T cell therapy in patients with ALL and DLBCL in 2017.
A series of preclinical and early clinical results suggest that CARs targeting B cell maturation antigen (BCMA), which is expressed in multiple myeloma, may emerge as the next successful clinical application of CAR T cell technology. Several groups, both academic and in the pharmaceutical industry, are developing BCMA-targeted CAR T cells, including the Nanjing Legend Biotech NCI, Kite, bluebird, and Novartis. Similarly, multiple antigens expressed by solid cancers are currently being targeted by CAR T cell approaches (Table 42.2 and Fig. 42.1). Early results from first-in-human studies targeting mesothelin (mesothelioma, pancreatic cancer, ovarian cancer) or EGFRvIII (glioblastoma) expressing malignancies have shown the safety of this approach. While available clinical efficacy results are far from those observed in B cell malignancies, the limited number of patients treated so far at the predicted clinically efficacious doses precludes drawing conclusions regarding the potential of CAR T cell therapies in solid tumors.
42.5.2 T Cell Receptor-Transduced T Cells
As mentioned earlier, investigators at the National Cancer Institute (NCI) demonstrated more than a decade ago that the adoptive transfer of in vitro expanded melanoma-reactive tumor-infiltrating lymphocytes (TILs) extracted from autologous fresh tumor samples induced tumor regression in 49–72% of patients with metastatic melanoma [16, 68]. However, this approach was only applicable to half of the patients with melanoma from whom TILs could be generated, with very limited success in other malignancies. This realization prompted the development of cell transfer studies in which T cells were genetically engineered to express TCRs specific against antigens expressed in a variety of human cancers. Early evidence that TCR-based gene therapy could induce significantly deep and durable responses in cancer was first provided by a clinical trial in which 2 of 13 patients experienced tumor regression upon adoptive transfer of autologous T cells engineered to express a MART-1-reactive TCR [17]. A follow-up study utilizing a higher avidity MART-1 TCR reported objective responses in 6 of 20 (30%) and in 3 of 16 (19%) patients treated with a murine-derived high-affinity TCR against human gp100 [24]. Unfortunately, severe on-target off-tumor toxicity, mostly affecting normal melanocytes in the skin, eye, and ear, highlighted the need to target antigens (nearly) absent in critical normal tissues. As mentioned earlier in this chapter, the adoptive transfer of T cells transduced with a MAGE-A3-reactive TCR led to the deaths due to unexpected of target cross-reactivity for two different TCRs and improved in vitro and in silico methods to predict cross-reactivity profiles for novel TCR lead structures are now available and will increase patient safety for novel TCR-engineered products. Thus far, only a limited number of studies exploring TCR T cell technology have shown consistent clinical activity. The most promising data have been generated by studies using TCR-engineered peripheral T cells targeting the cancer antigen New York esophageal squamous cell carcinoma 1 (NY-ESO-1). These trials therefore require special consideration.
42.5.2.1 Targeting NY-ESO-1
NY-ESO-1 is a CGA expressed in multiple tumors including in 10–50% of metastatic melanomas, lung, breast, and ovarian cancer as well as in 70–80% of synovial cell sarcomas [69, 70]. The first clinical study, using Gammaretrovirus to deliver a high-affinity TCR directed against an HLA-A*02-restricted NY-ESO-1 nonapeptide (residues 157–165) to autologous T cells, was published by investigators at the NCI in 2011 [6, 31]. Seventeen patients (6 with synovial cell sarcoma and 11 with melanoma) bearing tumors that stained strongly for NY-ESO-1 antigen expression (2 to 4+, >50% cells) were treated. Treatment consisted of lymphodepleting chemotherapy consisting of cyclophosphamide (60 mg/kg/d for 2 days) and fludarabine (25 mg/m2/d for 5 days) followed by NY-ESO-1 TCR-transduced T cells (median 5 × 1010; range, 1.6 to 130 × 109) and systemic IL-2. Nine patients responded, including two with melanoma that achieved a CR durable beyond 1 year and one with synovial sarcoma achieving a partial response (PR) that lasted 18 months. A recent update of this study provided data on additional 21 patients (12 with synovial sarcoma and 9 with melanoma) [6]. The ORR was 61% in synovial sarcoma, with estimated 3- and 5-year survival rates of 38% and 14%, respectively. In melanoma, the ORR was 55%, and the estimated 3- and 5-year survival rates were both 33% [6]. Overall, the toxicities observed in the trial were those expected from the lymphodepleting regimen and IL-2 therapy. These results validate NY-ESO-1 as an interesting cancer antigen for adoptively transferred T cell immunotherapy. Adaptimmune Therapeutics, a biotechnology company utilizing a proprietary SPEAR® (Specific Peptide Engineered Affinity Receptor) T cell engineering platform, has launched six clinical studies in HLA-A*02-positive patients with NY-ESO-1 expressing tumors. Fifty-three patients have been treated with NY-ESO-1c259SPEAR® as of January 2016, including 27 with multiple myeloma and 26 with a variety of solid tumors, including synovial sarcoma, melanoma, ovarian cancer, and NSCLC [71].
Of the trials in solid tumors, thus far, the most data is available from synovial sarcoma. Cohort 1 in the synovial study has completed accrual. Of the 12 patients with unresectable, metastatic, or recurrent synovial sarcoma, 6 achieved an objective response (ORR 50%). In the ovarian cancer and melanoma studies, six and four patients have been treated, respectively, but no objectives have been observed to date. It is worth noting that lymphodepletion in both these latter indications consisted exclusively of cyclophosphamide (i.e., no fludarabine), which might have contributed at least in part to the lack of objective responses. Both trials will continue treating patients using standardized NY-ESO-1 screening and fludarabine containing lymphodepleting regimens. Notably, NY-ESO-1c259 SPEAR™ T cells are able to persist over time, being detectable beyond 3 years post-infusion [71].
The TCR NY-ESO-1 paradigm was extended to multiple myeloma by investigators at the University of Pennsylvania using a lentivirus platform to force the expression on T cells of a TCR recognizing NY-ESO-1 [5]. Twenty patients with NY-ESO-1 expressing multiple myeloma received genetically engineered T cells 2 days after having undergone autologous stem cell transplant (SCT). NY-ESO-1 TCR-engineered T cells were safe, consistently trafficked to the bone marrow, and displayed extended persistence that correlated with clinical activity against multiple myeloma. The median progression-free survival (PFS) was 19.1 months, which suggests a potential role of this approach in patients with relapsed/refractory multiple myeloma. Moving forward, it will be important to determine the exact activity of NY-ESO-1 TCR T cells without the confounding effect of autologous SCT.
A recent publication reported that NY-ESO-1-engineered T cells show efficacy against disseminated neuroblastoma in xenograft mouse studies [72]. These nonclinical supportive data sets suggest that NY-ESO-1 may be a target with suitability beyond synovial sarcoma, malignant melanoma, and multiple myeloma.
Other Clinical TCR Targets
The activity of TCR T cell therapies beyond tumors expressing NY-ESO-1 has been infrequent and hampered by the limited persistence of TCR T cells, which may preclude the achievement of meaningful sustained responses, and fundamentally by tissue liabilities leading to untoward toxicity. Multiple cancer-associated antigens expressed at low levels in normal tissues such as CG antigens, CEA, and Her2 are currently being targeted by TCR-engineered T cells (Table 42.1). More clinical data from a next wave of TCR-engineered T cells targeting a variety of different antigens will become available in the next 2–3 years and contribute much to our understanding about the best targets and TCRs for the treatment of patients with cancer (Table 42.4).
Table 42.4
TCR T cell product in the clinic