15 Total Body Irradiation
History
Only a decade after Roentgen described the “x-ray,” German biophysical engineer Friedrich J. Dessauer first described a “new technique of radiotherapy” that involved homogenous irradiation of the entire body. In his initial report describing the technique in 1905, he proposed irradiating a supine patient using three simultaneously active low-voltage Roentgen ray sources (Fig. 15-1).1,2 In 1907, Aladár Elfer, a medical professor in Hungary, reported his experience using a TBI technique that spared the head in three patients with leukemia.3 Although there is a paucity of data regarding the early use of the technique, some have speculated that untoward hematologic toxicity probably limited its application.4
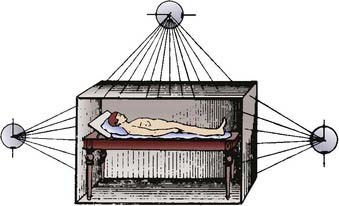
FIGURE 15-1 • Diagram demonstrating the total body irradiation technique proposed by Dessauer in 1905.
Early success using TBI to treat hematopoietic and lymphoid malignancies in Europe (there named the Teschendorf method) prompted development of the technique in the United States.5–7 Arthur C. Heublein, in collaboration with Gioacchino Failla, is credited with the development of the first TBI unit in North America, located at Memorial Hospital in New York City. In the United States, the technique became known as Heublein therapy.8 A specially constructed treatment ward was designed to treat four patients at an extended distance (5-7 meters) simultaneously at an exposure rate of 0.7 R per hour for approximately 20 hours per day, typically over 1 to 2 weeks, using a 185 kV x-ray tube at 3 mA, with a 2-mm copper filter. The goal was to deliver 25% of erythema dose (750 R). Remarkably, in Heublein’s initial report, no hematopoietic toxicity was noted with this treatment schedule. Of 12 patients with advanced lymphomas and leukemias, 7 (58%) demonstrated some form of improvement after treatment; 2 of 8 patients (25%) with metastatic breast, melanoma, and kidney cancers were also noted to demonstrate some form of improvement.9,10 A later report of the experience with 270 cancer patients from Memorial Hospital treated with TBI between 1931 and 1940 confirmed that the technique was more successful in patients with hematopoietic and lymphoid malignancies, compared with patients with carcinomas or sarcomas, for whom it was ineffective. The authors emphasized that the technique was safe if doses were prescribed cautiously. They did not recommend exposures greater than 300 R, and noted hematopoietic and gastrointestinal toxicity with exposures as low as 50 to 100 R.8
In the early 1940s, World War II prompted the Manhattan Project, an initiative to develop nuclear weapons. Part of this endeavor sponsored research into the human biologic response to ionizing radiation, including TBI. The military’s interest in TBI was primarily to help understand human tolerance for radiation exposure during occupational duties and warfare, and to develop radiation biodosimetric assays. Several research studies coordinated through the Manhattan Project were initiated in patients with advanced malignancies,11–13 as well as in patients with benign diseases.13 For example, studies of dose escalation, radiation biologic dosimetry, and cognitive and psychomotor function were carried out at M.D. Anderson Hospital for Cancer Research.14 A detailed report of 30 patients treated at the maximum exposure level (200 R) in the initial study concluded that side effects primarily consisted of nausea, vomiting, and myelosuppression, and that intervention was necessary in 10% of patients treated with this dose of TBI.15 At Baylor University College of Medicine, studies using 25 to 250 R of TBI with 250 kV to 2 MV energy machines were performed to find a biologic dosimeter, as well as to study acute effects of radiotherapy.16 The Sloan-Kettering Institute for Cancer Research also participated in similar studies of at least 20 patients, although the results were never published. The military conducted similar studies at the Naval Hospital in Bethesda, Maryland, and reported palliation of patients with radiosensitive diseases treated with fractionated TBI.17
The most recent Department of Defense–sponsored research study of TBI was at the University of Cincinnati and focused on identifying biochemical markers in the urine that predicted response to TBI. Later, studies of the neuropsychiatric effects of TBI were initiated. Ultimately, only results regarding the palliation of advanced malignancies were reported.18 Patients with advanced metastatic radioresistant malignancies for whom chemotherapy was unavailable were often treated with TBI in the absence of any clear anticipated benefit. Patients treated with TBI in this manner were included in research studies, often without consenting to participate. The ethics of this practice were called into question by a report written in 1995 by the United States Department of Energy’s Advisory Committee on Human Radiation Experiments,19 which could be a contributor to the public’s uneasiness with radiation in general.20
TBI was used not only in malignant diseases; it was considered the critical immunosuppressant in the first successful solid organ transplant. In 1959, a kidney was successfully transplanted between dizygotic twins after TBI to 450 R to the recipient.21 Around the same time in France, successful kidney transplants after TBI were being reported.22,23 Of the first seven patients who underwent kidney transplant following TBI or pharmacologic immunosuppression worldwide between 1959 and 1962, the two who did not experience kidney failure were treated with TBI alone (without chemical immunosuppression) prior to transplant, and each survived in excess of 20 years after transplant.21–23 However, successful preclinical studies with pharmacologic therapy prompted the use of chemical immunosuppressants (corticosteroids, 6-mercaptopurine, and azathioprine) for solid organ transplantation after 1963.24
With an increased understanding of the human response to TBI, and a rapidly growing body of preclinical in vivo studies of TBI, therapeutic protocols were developed to maximize benefit in patients with malignant diseases. In 1957, Nobel laureate E. Donnall Thomas first reported the use of bone marrow infusion in humans following whole-body irradiation or chemotherapy,25 and less than one year later he published his experience using TBI with exposures up to 600 R, followed by bone marrow transplantation.26 In the series of the first five patients with leukemia treated with TBI, who then received intravenous infusion of normal donor marrow, Thomas et al. noted the difficulty of acute myelosuppression and resultant hemorrhage and infection during the period leading up to engraftment. The report also comments that low dose rates (delivery over 2-3 days) appeared preferable to higher dose rates, for metabolic and immunologic reasons. In addition, patients receiving 200 to 300 R fared better than those receiving 400 to 600 R. The problem of delivering an adequately homogenous dose was raised, and suggestions to use higher-energy photons were proposed.26 Thomas et al. later reported on syngeneic bone marrow transplantation in two children after 850 to 1140 R was delivered in a single fraction over 22 to 25 hours, using 60Co sources. The authors concluded that 1000 R of TBI did not produce “troublesome” acute radiation sickness, and produced remission of leukemia, but not cure.27 The first report of successful cure of a patient with leukemia with allogeneic transplantation after TBI was reported in 1969. The technique involved opposed 60Co sources, which operated at 5.8 R per minute, to a total exposure of 1620 R, calculated to be 954 rad at midline. With appropriate supportive care, no major acute radiation sickness was noted, but the patient died of overwhelming cytomegalovirus (CMV) infection, without evidence of leukemia.28
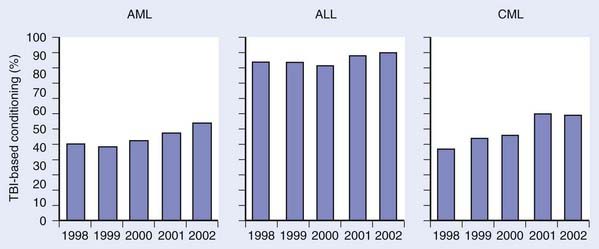
Over the next several years, techniques of combining chemotherapy and TBI were developed and refined, with promising results.29,30 Success in the treatment of advanced leukemias and severe aplastic anemia (AA) was achieved. Departure from the use of TBI alone was primarily fostered by the development of more effective cytotoxic chemotherapeutics and immunologic therapies, which, when combined with TBI, yielded fewer leukemic recurrences.29 Although the use of TBI without HSCT has largely been abandoned, primarily for fear of inducing secondary malignant neoplasms (SMNs) and limiting later therapeutic options, some question the validity of this fear and still contend that very low-dose TBI (1.5-2 Gy in 10-20 fractions over several weeks) is a viable option for upfront therapy in advanced indolent lymphomas.31 Fig. 15-2 demonstrates how the use of TBI during HSCT has remained constant or increased during preceding years.32
Background on Hematopoietic Stem Cell Transplant
HSCT has evolved into a highly complex clinical discipline, firmly rooted in immune system and cancer biology, the details of which are beyond the scope of this chapter. When HSCT was initially performed (see “History”), bone marrow was extracted from the donor, filtered, and infused intravenously to the recipient. Later, peripheral blood stem cells, instead of bone marrow, were collected from the donor, and found to be an alternative to bone marrow grafts. For this reason, “bone marrow transplants” are now more appropriately termed hematopoietic stem cell transplants, because the critical component of the graft is the hematopoietic stem cell (HSC), independent of the source. Oftentimes, peripheral blood stem cells are “mobilized” from the donor using hematopoietic growth stimulating factors, and removed from the donor by apheresis. In addition, recent success using HSCs derived from human umbilical cord blood has been described. Depending on the source of HSCs, various postcollection processing measures (cell selection and depletion) may be undertaken to optimize the outcome of the transplant.
When transplant occurs between different individuals, the hematopoietic graft is said to be allogeneic. This is in contradistinction to reinfusion of native HSCs back into the donor in an “autologous” transplant, more appropriately termed an autoplant, because nothing is being transferred between different individuals.33 A rare but alternative situation is of a patient with a genetically identical twin in the case of a “syngeneic” transplant. Of these three methods of HSCT, autologous and syngeneic transplant are generally associated with less risk because issues related to immunocompatibility are minimized. Allogeneic transplants requiring the “matching” of donor and recipient are typically carried out through identification of human leukocyte antigen compatibility. The donor may be related to the recipient, or identified through registries of volunteers, such as the National Marrow Donor Program.
Prior to undergoing HSCT, most patients require intensive antineoplastic or immunosuppressive therapy in preparation for HSCT, often referred to as conditioning. Conditioning can involve cytotoxic chemotherapy, immunosuppressants, antibody therapy, or radiation therapy. The nature of the conditioning regimen can be referred to as high or reduced intensity, myeloablative, submyeloablative, or nonmyeloablative, to carry out conventional or mini transplants.33 Although agreed-upon formal definitions of these regimens do not exist,34 the goal of high-intensity, myeloablative, or conventional transplant is to completely eliminate the recipient’s native HSC compartment, which necessitates HSCT (autologous or allogeneic) for survival. High-intensity, myeloablative, or conventional transplants may or may not involve TBI, to high doses (>5 Gy in a single fraction, >8 Gy in multiple fractions). Reduced-intensity, nonmyeloablative, or mini-transplant conditioning regimens are often used for older patients or for those with medical problems for whom a high-intensity, myeloablative, or conventional transplant would cause excessive morbidity or mortality, and may or may not involve TBI to lower doses. During and immediately after conditioning, the transplant recipient is at significant risk of infections and other hematologic complications. For this reason, the supportive care of HSCT recipients is very complex, and should only be undertaken in specialized facilities. Nonetheless, some groups have developed reduced-intensity and myeloablative HSCT regimens, including TBI, which have been safely undertaken on an outpatient basis.35,36
According to data summarized by the Center for International Blood and Marrow Transplant Research (CIBMTR) in 2005, the diseases most commonly treated with HSCT are (in decreasing order of frequency): multiple myeloma (MM); non-Hodgkin lymphoma (NHL); acute myelogenous leukemia (AML); Hodgkin disease (HD); acute lymphoid leukemia (ALL); myelodysplastic and myeloproliferative disease (MDS); chronic myelogenous leukemia (CML); AA, and various other leukemias, cancers, and nonmalignant diseases.37 The 2009 National Comprehensive Cancer Network (NCCN) guidelines for therapy indicate that allogeneic or autologous HSCT may be a treatment option for AML, MM, MDS, CML, HD, and NHL, depending on the clinical situation. Detailed discussion of these diseases and their management is beyond the scope of this chapter, and the reader is referred to appropriate chapters in this text and others for further review.38–41 The role of TBI in nonmalignant diseases is discussed further below.
Radiobiology
Preclinical studies have helped define some of the fundamental radiobiologic properties of the normal lymphocytes. The D0 of normal lymphocytes has been reported to be 0.5 to 1.4 Gy,42–45 depending on the in vitro or in vivo model used to calculate this parameter. This D0 suggests that normal lymphocytes cells are very sensitive to ionizing radiation. A very small shoulder on the radiation cell survival curve has been noted,46,47 suggesting little repair between fractions of radiation. Clinical data have revealed similar findings in patients undergoing hyperfractionated TBI (HTBI), with lymphocyte survival demonstrating an effective D0 of 3.8 Gy according to one study.48 Other radiobiologic phenomena have been ill-defined in other normal hematopoietic cells. Radiobiologically relevant levels of hypoxia are unlikely in the hematopoietic compartment. Repopulation is not likely to influence hematopoietic cell survival, given the short duration of most TBI regimens (1-5 days), although, given the variable lifespan of leukocytes (days to years), it may be of some relevance. Redistribution appears to be of significance, given the time scale for TBI; however, this has been difficult to assess.49
The radiobiology of malignant hematopoietic cells has been described. The D0 of leukemic cells generally ranges between 0.8 and 1.5 Gy, however, compared with normal hematopoietic cells, a wider range of radiosensitivities have been described.50–61 Many have cited the technical nuances and variations in assay technique for this great range.49,62 Similar to normal hematopoietic cells, their malignant counterparts are thought to demonstrate little sublethal damage repair,63–67 although split-dose and low-dose-rate experiments have demonstrated the capacity for leukemic cells to repair radiation-induced damage.55,58,68,69 Generally, leukemic cells are believed to have a cell survival curve with minimal or no shoulder, although this varies across cell types and cell lines.47,51,54 For example, Cosset and colleagues summarized preclinical and clinical findings, concluding that AML demonstrates little repair, whereas CML does demonstrate repair, and ALL, myeloma, and lymphomas have not been well studied but appear to demonstrate a wide range of repair capacity.62,70 Similar to normal hematopoietic cells, reoxygenation is unlikely to be radiobiologically relevant to malignant hematopoietic cells during TBI. Redistribution and repopulation, however, may be relevant, but have not been systematically studied.
In vivo preclinical research laid the foundation for the first successful HSCT in humans. Studies in rats,71 dogs,72 and nonhuman primates73 demonstrated that reconstitution of the hematopoietic system was possible after TBI with supralethal doses of radiation. Later work in animals revealed that delivering TBI in several fractions required a higher total dose relative to the biologically isoeffective dose given in a single fraction.74–76 Another model demonstrated no significant difference in the effect of a low-dose-rate (0.04 Gy/min) single fraction of TBI compared with a hyperfractionated course of TBI given thrice daily to the same total dose.77
Although the hematopoietic system is the target of TBI, normal tissues effectively limit the dose that can be safely delivered. The sparing of normal tissues with fractionated TBI was proposed by Peters and colleagues,63,64 and subsequently supported by preclinical data in mice78,79 and dogs80 showing less lung injury with fractionated TBI regimens.
Immediate Toxicity and Management
Although a good deal of what has been learned about the acute in vivo biologic effects of TBI is derived from laboratory-based animal studies, whole-body irradiation also has been studied in people exposed during accidental or wartime nuclear events.81,82 These large-scale studies are valuable because they deal with apparently normal subjects; however, the retrospective nature limits the quality of the data. The reader is referred to several excellent texts for a review of the acute and fatal radiation syndromes (gastrointestinal, hematopoietic, and cerebrovascular) that can be caused by TBI in an uncontrolled setting.83–85
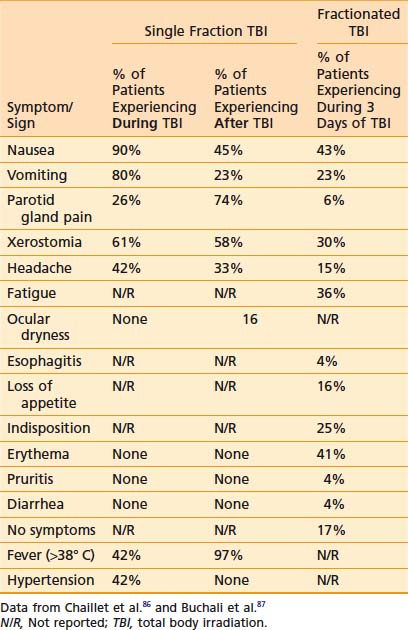
Acute side effects of therapeutic TBI can be difficult to distinguish from other HSCT-related morbidity. However, Chaillet and colleagues conducted a very informative prospective clinical study of the symptoms and signs that occur in patients after TBI, prior to the initiation of any other HSCT related therapy.86 Thirty-one patients between 4.5 and 55 years of age were treated using parallel-opposed anteroposterior (AP) 18 MV photons from a linear accelerator (linac). Shielding was used to limit the lung dose to 8 Gy. A total dose of 10 Gy was given as a “single dose” as six discrete fractions of 1.6 Gy, each given over 15 minutes, with a 30-minute break between fractions, for a mean dose rate of 0.04 Gy per minute and instantaneous dose rate of 0.11 to 0.12 Gy per minute. Symptoms and signs were assessed regularly during the 4 hour TBI, and for 20 hours after the completion of TBI. Antiemetics, but no chemotherapy or steroids, were given prior to the start of TBI. Table 15-1 displays the symptoms and signs experienced by patients during the 4 hours of TBI, and within 24 hours of starting TBI. Fever was a very common finding, and a maximum of 40.8° C was noted in one patient. Tachycardia frequently paralleled febrile episodes; a maximum rate of 130 beats per minute was noted. Drowsiness was noted only in patients who received sedating antiemetics. Parotid gland pain was common, and bilateral parotid swelling was noted in 29% of patients. Marked lacrimation was noted in 6% of patients, whereas 16% of patients experienced ocular dryness. Two patients experienced mild conjunctival edema. Hypertension was noted only during TBI. The results of a similar study conducted by Buchali and colleagues of patients who were treated with a fractionated course of TBI delivered mostly to a total dose of 12 Gy using 2 Gy per fraction, twice daily, 8 hours apart, with lung doses limited to 10 Gy, is also summarized in Table 15-1.87
A prospective clinical study showed that fractionation of TBI can reduce acute nausea, vomiting, mucositis, diarrhea, and parotitis, although these differences were not statistically significant. Late cutaneous eruptions were more common in patients undergoing fractionated TBI, although this was not statistically significant. This same study, which randomized patients to high- or low-dose-rate TBI, revealed no differences in the acute toxicities mentioned when comparing dose rate.88 Another randomized controlled trial (RCT) reported that fractionating TBI revealed “no apparent difference in acute toxicity” compared with single-fraction TBI, with both regimens being “well-tolerated.”89
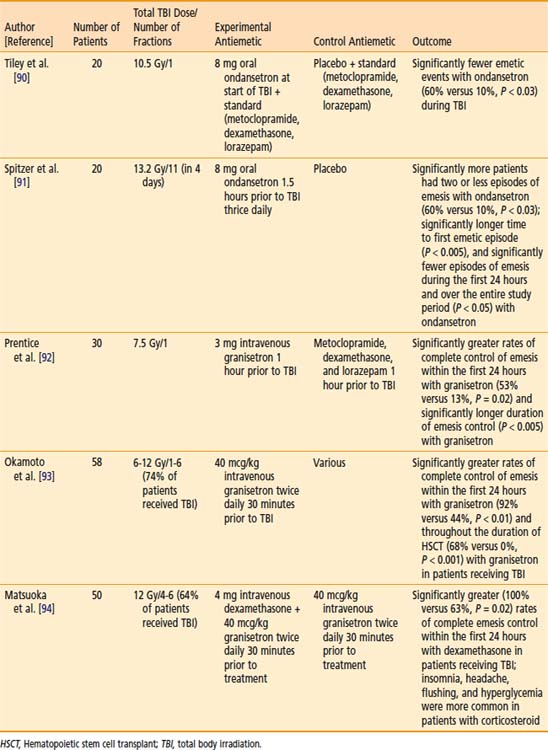
Older studies cite nausea and vomiting as frequent side effects of TBI. These symptoms have been substantially minimized with the advent of more effective antiemetics, such as the 5-hydroxytryptamine (serotonin) receptor-3 (5HT-3) antagonists. Several small, high-quality controlled clinical studies support the prophylactic use of 5HT-3 antagonists to reduce nausea and vomiting during TBI, and are summarized in Table 15-2.90–94 The use of corticosteroids in conjunction with 5-HT3 antagonists is supported by a trial listed in Table 15-2; however, given the toxicity associated with this approach, consensus regarding routine administration in conjunction with TBI is lacking.95–97 Of note, less nausea and vomiting has been noted in myeloablative conditioning regimens involving TBI, compared with those which use chemotherapy alone, even with modern antiemetics.98
Table 15-2 Randomized Controlled Trials of Prophylactic Antiemetics in Patients Undergoing Total Body Irradiation
Oral mucositis is a side effect of TBI in up to 75% of patients undergoing myeloablative TBI, causing mouth pain, odynophagia, necessitating intensive supportive care such as total parenteral nutrition and opioid analgesics.99 In one study, intensive dental hygiene conferred a reduction in the rate of moderate and severe mucositis, although the authors felt it to be clinically insignificant.100 Topical oral agents, such as chlorhexidine digluconate and neutral calcium phosphate in conjunction with topical fluoride treatments, can decrease the duration and severity of oral mucositis as well as pain and the need for opioid analgesics.101–103 Similarly, prophylactic oral sucralfate and clarithromycin have reduced moderate and severe oral mucositis rates.104,105 When given prophylactically, one study showed that amifostine limited the duration of mucositis, with an associated decrease in the rate of moderate and severe infections, with no effect on HSCT outcome.106 One study noted that short-term intravenous recombinant granulocyte-macrophage–colony stimulating factor decreased rates of moderate to severe mucositis,107 whereas another found no effect when this agent was delivered topically.108 Recently, Spielberger et al. reported the results of a trial of the recombinant human keratinocyte growth factor, palifermin, given before and after conditioning with 12 Gy of fractionated TBI. Palifermin reduced the rate and duration of moderate and severe mucositis by 35% and 3 days, respectively, and decreased mouth and throat pain, as reflected in reduced morphine usage and decreased the need for total parenteral nutrition (by 24%).109 This study dealt only with patients undergoing autologous HSCT; however, in the setting of TBI for allogeneic HSCT, palifermin may also confer a protective effect on the mucosa, although this has not been studied in an RCT.110
Skin erythema may also be noted toward the end of a course of TBI; desquamation is rare. Hyperpigmentation may be noted in the long term. Alopecia typically occurs 7 to 14 days after TBI is complete, and hair typically returns 3 to 6 months after treatment.111 Changes in the color or texture of regrown hair have been noted by some. Of note, myeloablative conditioning regimens using chemotherapy alone have noted a significantly higher incidence of permanent alopecia.112
Later Toxicity and Management
As previously mentioned (see “Radiobiology”), the hematopoietic system is particularly sensitive to TBI, and lymphopenia is often seen with doses of 0.5 Gy, and can be seen with doses of 0.3 Gy. Lymphopenia is typically followed by neutropenia, thrombocytopenia, and finally anemia. Soon after a TBI dose of 4 to 6 Gy, lymphocytosis can be seen, but typically is followed by neutropenia within 1 week. Neutrophils fall to their minimum 3 to 4 weeks after TBI (Fig. 15-3).113 Regeneration of the HSC compartment depends on the total dose used, because higher doses cause more rapid myelosuppression, of greater duration. Administration of hematopoietic growth factors after TBI have the theoretical potential to alter hematopoietic system reconstitution, although reports in the setting of allogeneic HSCT have demonstrated an increased risk of GVHD and compromised survival,114 and therefore routine use is controversial.115,116 Of note, hematopoietic growth factors have only been used in the post-TBI period, given the concerns raised by a trial in lung cancer, in which growth factors increased pulmonary toxicity and thrombocytopenia when given concurrent with chemoradiation therapy.117
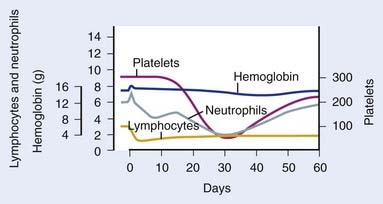
FIGURE 15-3 • Representative hematologic response to total body irradiation, given as a single fraction of 200 cGy on day 0.
(From Andrews GA: Radiation accidents and their management, Radiat Res Suppl 7:390–397, 1967.)
As noted previously, salivary glands frequently are affected by TBI. Although acute parotitis is typically self-limited and can be managed with anti-inflammatory medicines, long-term salivary gland dysfunction can result in xerostomia, which may lead to dental caries. In a study of children who underwent allogeneic HSCT, the risk of developing impaired salivary function was 22% in those who receive TBI as part of conditioning versus 1% in those who did not.118 Studies have shown that salivary flow can improve up to 1 year after the completion of TBI.119 Fractionating TBI was shown to reduce salivary dysfunction by 54% in one study.120 Myeloablative conditioning regimens with and without TBI have been associated with tooth development abnormalities in children.120–122 In one series, myeloablative conditioning regimens using chemotherapy alone were associated with significantly higher rates of tooth developmental abnormalities than those involving TBI, although rates of salivary gland dysfunction were highest amongst the patients treated with single-fraction TBI.123 Because of the increased risk of oral pathologic conditions associated with TBI, careful pretransplant evaluation by a dental specialist is recommended to minimize the risk of serious morbidity.124 Pilocarpine has been noted to help relieve symptoms of xerostomia in patients treated with TBI.125
The major dose-limiting toxicity of TBI is pneumonopathy, which can manifest early as pneumonitis and later as pulmonary fibrosis. In the setting of HSCT, radiation pneumonopathy can be difficult to distinguish from other causes of pathologic conditions of the lung; moreover, lung damage is likely multifactorial, with risk of acute lung complications estimated to be 30% to 60%, depending on factors like infection, conditioning regimen, GVHD, age, and diagnosis (see Peters and Afessa126 for review). Likewise, late pneumopathy occurs in 10% to 26% of patients, and is associated with underlying lung dysfunction, type of conditioning regimen, acute and chronic GVHD and prophylaxis, donor and recipient age and immunocompatibility, stage of disease, and genetic predisposition (see Patriarca et al.127 for review). TBI has been shown to be a risk factor for the idiopathic pneumonia syndrome128 as well as diffuse alveolar hemorrhage.129 Although rates of pneumonopathy in patients receiving TBI vary widely (10%-84%),130 some series have reported pneumonitis in up to 20% of HSCT patients who never received TBI.131 In the modern era, with appropriate TBI techniques, the risk of pneumonopathy in patients treated with TBI may not be increased at all.132 Nevertheless, the significance of the problem is clear, given that mortality related to interstitial pneumonitis in patients treated with TBI can be 60% to 80%.131,133,134
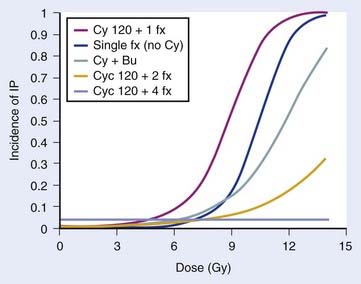
Several TBI-specific factors (total dose, fractionation, dose rate, and the use of lung shielding) have been shown to have significant bearing on the development of pulmonary complications. The total dose used during TBI has frequently been cited as a major factor influencing lung complications.130,135,136 In two prospective RCTs using 12 Gy versus 15.75 Gy, higher rates of mortality were noted within the first 6 months in patients treated with 15.75 Gy, although pulmonary complications were not specifically cited as the excess cause of deaths.137–140 In a retrospective dosimetric study, mean lung dose larger than 9.4 Gy was found to be an independent predictor for lethal pulmonary complications in patients receiving TBI to a total dose of 10 Gy in three daily fractions, at 0.055 Gy per minute using parallel opposed lateral fields.141 Two RCTs have demonstrated that fractionated TBI can reduce pneumonitis, compared with single-fraction TBI, although only one study was statistically significant.89,142,143 A retrospective study found no difference in pneumonitis rates when comparing a single fraction of 6 Gy and three daily fractions of 3.33 Gy, suggesting that total doses smaller than 10 Gy may not require fractionation to prevent toxicity, although no randomized data support this.144 The necessity of hyperfractionation to prevent lung toxicity is unclear: a comparison of two prospective single-arm trials at the same institution revealed that conventional fractionation given with AP fields and lung blocks to a total dose of 12 Gy in daily 3-Gy fractions may not be any different than HTBI given twice daily with 1.7 Gy per fraction to a total dose of 10.2 Gy for 3 days, using parallel opposed lateral fields and no blocks.145 Sampath et al. recently reviewed 26 studies involving 1096 patients to create a dose-response model to predict risk of pneumonitis from TBI, taking other factors into consideration. Although unable to estimate the risk of pneumonitis for hyperfractionated regimens, they were able to determine the effect of fractionation and cyclophosphamide (CY) and busulfan (Bu) on the risk of developing pneumonitis, in a dose-response model, as seen in Fig. 15-4.146 Pneumonitis rates were not significantly different in a trial that randomized patients to high- or low-dose-rate TBI.88 However, there is an abundance of retrospective clinical data suggesting that lowering the dose rate (<0.025-0.09 Gy/min) does decrease the likelihood of pulmonary complications,130,136,140,147,148 especially if TBI is delivered as a single fraction.149 If TBI is fractionated, some report that low dose rate (<0.069 Gy/min) is not necessary,134 although others found a beneficial effect.150,151 Studies of patients receiving fractionated TBI with lung shielding have demonstrated a reduction in pneumonopathy152,153; however, one RCT found no difference in pneumonopathy if the shielding allowed a lung dose of either 6 or 8 Gy in a single fraction.154
Pulmonary function tests (PFTs; spirometry and diffusion capacity) are often helpful in the assessment of patients with pulmonary symptoms or radiographic abnormalities. Studies of PFTs in patients treated with HSCT have demonstrated a deleterious effect on spirometry and diffusion capacity, which often resolves in the absence of other complicating factors,155–157 and is related to TBI dose.158 A retrospective study found that lung shielding had a small but significantly beneficial effect on PFTs one year after HSCT, especially in patients with abnormal function prior to HSCT.159 In one study, Bu, but not TBI, was not associated with a negative effect on PFTs.160 There is no evidence that PFTs improve pulmonary outcome after HSCT in adulthood, and for this reason, they are not recommended.161 However, some groups recommend baseline PFTs as part of long-term follow-up care for children treated with TBI.162 Counseling patients regarding smoking cessation is of critical importance in all patients, especially those who are at increased risk of developing lung injury. In the event of acute pneumonitis, high-dose steroids (30-60 mg of prednisone/day) typically alleviate symptoms within 24 to 48 hours.
Cardiovascular toxicity as a result of HSCT has been relatively rare, given the stringent selection criteria for patients treated with this aggressive therapeutic modality. Nevertheless, among patients who survive HSCT, cardiac events were responsible for 2.4% and 3.0% of deaths in patients that underwent autologous and allogeneic HSCT, respectively, which represents a greater than expected occurrence in some patients.163,164 Most of the recent literature has identified no association between TBI and development of cardiovascular disease.165,166 Several detailed prospective analyses using plasma cardiac troponin and brain natriuretic peptide levels, electrocardiography and echocardiography revealed no evidence of cardiac dysfunction in previously healthy individuals treated with TBI.167,168 However, a prospective study of children who underwent allogeneic HSCT found a 12% and 26% cumulative incidence of abnormalities in ejection fraction (<30%) on echocardiography prior to and five years after HSCT, respectively. This study revealed that TBI was associated with abnormalities in cardiac function on univariate analysis, but not multivariate analysis; the five-year cumulative incidence of cardiac abnormality was 26% in children treated with TBI with prior anthracycline therapy and 2% in children treated with TBI without prior anthracycline therapy.169 For these reasons, it appears prudent to screen patients who have undergone HSCT for cardiovascular disease or risk factors, whether or not this involved TBI, to minimize late morbidity and mortality.
Hepatotoxicity from TBI manifests primarily as veno-occlusive disease (VOD), also known as sinusoidal obstructive syndrome, of the liver. This clinicopathologic phenomenon was first described in 1977 by Shulman et al.,170 who noted the onset of weight gain from ascites, painful hepatomegaly, and jaundice from centrilobular liver acinus necrosis 1 to 4 weeks after HSCT. Overall, up to 70% of patients that undergo HSCT can be affected by VOD. TBI, along with many other risk factors, has been implicated in the development of VOD (see Wadleigh et al.171 for review). Notably, a RCT and meta-analysis found that patients treated with Bu instead of TBI were significantly more likely to develop VOD.172,173 Two RCTs have concluded that fractionated TBI reduces the incidence of VOD compared with single-dose TBI.142,143 Another RCT found no difference in the rate of VOD when the dose rate was either 0.06 Gy per minute or 0.15 Gy per minute,88 although a retrospective study of single-dose TBI found that dose rates of 0.07 Gy per minute were associated with less VOD than dose rates of 0.18 to 1.2 Gy per minute.174 TBI dose greater than 13.2 Gy has been reported to be associated with higher rates of VOD on univariate analysis,175 although dose greater than 12 Gy was not associated with VOD in another retrospective study.176 Lawton et al. reported a non–statistically significant 10% decrease in the rate of fatal VOD in patients treated with TBI as part of HSCT, when a 10% attenuation liver block was employed.177 Ursodeoxycholic acid was effective in preventing VOD in a RCT of patients undergoing TBI followed by HSCT,178 although another RCT did not support this finding.179 Reduced-intensity conditioning regimens may also prevent VOD. Treatment of VOD can include the fibrinolytic antithrombotic agent, defibrotide; decompressing the sinusoids by a transjugular intrahepatic portosystemic shunt and liver transplantation are more invasive options for management of severe disease.180
Cataracts are one of the most common complications of TBI. Patients may present with painless vision loss, and may be noted to have opacification of the lens on examination. In one series of patients treated with TBI in one or two fractions, severe visual impairment was noted in approximately half of patients.181 This problem has been noted to arise in a large proportion of patients treated with TBI, depending on total dose, fractionation, and dose-rate used. When considering risk factors associated with HSCT, steroid use,182–184 prior cranial irradiation,185,186 and the development of GVHD184 have been shown to predispose to cataractogenesis, whereas heparin use appears to be protective.187 Delivering TBI in a single fraction is the single biggest risk factor for developing a cataract after HSCT.187–192 High dose rate (>0.035-0.048 Gy/min) TBI also appears to increase the risk of cataract formation.185,187,190,193 A prospective study that randomized patients to high- or low-dose-rate TBI found that the incidence of cataract 5 years after treatment was 12% and 34% in the low- and high-dose-rate arms, with 13% and 39% of the cataracts occurring in patients who received fractionated or single-dose TBI, respectively.194 Kal and van Kempen-Harteveld recently reviewed the subject of TBI-induced cataracts and concluded that a biologically equivalent dose of 40 Gy yields a 10% chance of developing cataracts, using linear-quadratic modeling that included corrections for dose rate, with an α/β of 0.65 for late effects on the lens. On the basis of this, they suggest considering lens shielding for single-fraction TBI regimens,195 although this is controversial in the setting of malignant disease.196 Given the frequency of cataracts occurring after TBI, patients should be monitored for the development of this complication.197 Management of cataracts that impair vision or degrade quality of life may involve phacoemulsification or extraction; recent data suggest these procedures are safe, with an adverse event rate of 0.1% for experienced surgeons,198 and effective, with a 90% chance of 20/40 vision postoperatively.199
Kidney dysfunction occurs in approximately 17% of survivors of HSCT,200 and can manifest in a number of ways, with the most- to least-frequent syndromes being idiopathic chronic kidney disease, nephrotic syndrome, thrombotic microangiopathy (thrombotic thrombocytopenic purpura and hemolytic uremic syndrome), and acute renal failure (see Hingorani201 for review). The syndrome best associated with TBI is thrombotic microangiopathy, which can manifest as nephritis, hypertension, proteinuria, or anemia 6 to 12 months after HSCT. HSCT-related risk factors for nephropathy can include GVHD, infections with CMV or BK virus, nephrotoxic medications like cytotoxic chemotherapy (cytarabine, CY, ifosfamide, cisplatin, retinoic acid, carmustine, actinomycin D, melphalan), antibiotics (acyclovir, ganciclovir, foscarnet, vancomycin, amphotericin, aminoglycosides), and immunosuppressants (cyclosporine, tacrolimus, methotrexate). It is worth noting that the larger and more contemporary studies of chronic kidney disease after HSCT, have not demonstrated an association with TBI.202–206 Total dose has been implicated as the most important in predicting renal morbidity from TBI; a retrospective study found that GVHD and high dose (13.5 Gy) was associated with elevated serum creatinine.207 A prospective evaluation of renal function using radioisotopes found that early nephropathy was associated with age younger than 40 years, use of kidney blocks (possibly related to nephrotoxic contrast media given during simulation), and nephrotoxic drug use, while late nephropathy was associated with nephrotoxic drug used, but not TBI dose.208 The benefit of kidney shielding has been assessed in two retrospective studies, which both demonstrated significant improvement in long-term kidney function, with no evidence of dysfunction when the hyperfractionated dose was limited to 9.8 to 10 Gy.209,210 Two recent dose-effect modeling studies have demonstrated that nephropathy is unlikely after a biologically equivalent total dose of 16 Gy (calculated using a linear quadratic model, with corrections for dose rate and an α/β of 2.5 Gy) and that fractionating TBI and delivering a low dose rate (<0.10 Gy/min) prevent kidney dysfunction.195,211 Monitoring blood chemistry and counts, as well as blood pressure and urine studies are advised given the prevalence of kidney disease after HSCT. Although there are no proven therapies to treat HSCT-related nephropathy, medical therapies (antihypertensives, corticosteroids), plasma exchange, hemodialysis and renal transplant, and are possible options for management (see Hingorani201 for review). The angiotensin-converting enzyme inhibitor captopril is known to prevent chronic nephropathy from diabetes212 and was been hypothesized to reduce the risk of radiation nephropathy from TBI in preclinical studies.213,214 A small RCT of captopril to mitigate chronic renal failure in patients undergoing 12 Gy of fractionated TBI (with kidney dose of 9.8 Gy) prior to HSCT revealed less nephropathy; however, this was not statistically significant.215
Primary hypothyroidism is the most common endocrinopathy following TBI with an incidence of approximately 25% after fractionated therapy.216 The incidence is significantly higher in series of single-dose TBI and is known to increase with longer follow-up. It has been suggested that adults have a lower risk of hypothyroidism than children and that some patients develop only transient hypothyroidism. This condition is easily managed with thyroid hormone replacement. Following TBI, central hypothyroidism caused by thyrotropin (thyroid-stimulating hormone) deficiency is rare, as is deficiency of corticotropin (adrenocorticotrophic hormone), and hypogonadotropic hypogonadism.
Impaired growth and loss of adult height has been described as a consequence of TBI during childhood. This effect is multifactorial because radiation can cause poor nutritional conditions, skeletal dysplasia, and also temporary or permanent growth hormone (GH) deficiency. The effects of TBI on growth are inversely related to patient age at the time of treatment. Both the total dose and dose per fraction of TBI have shown to be related to the risk of growth impairment. These factors are also important when considering GH replacement for children proven to have deficiency. Sanders et al. observed that GH replacement was not effective after single-dose TBI or when TBI was delivered after age 10 years.217
Typical TBI regimens allow preservation of testicular Leydig cell function and therefore normal testosterone levels. However, almost all males will develop azoospermia as a result of germ cell irradiation during TBI. Although male patients are universally counseled to expect infertility, there are rare reports of spermatogenic activity in men many years after exposure to TBI during childhood. Age plays an even more important role in female fertility following TBI. Although approximately half of prepubescent girls who receive TBI will undergo spontaneous puberty, many will experience premature menopause in their twenties or thirties.218 Ovarian failure is expected in virtually all females receiving TBI after age 10 years. For the small percentage of women who become pregnant after TBI, some investigators have reported an increased risk for spontaneous abortion, whereas others do not.
Other endocrine and metabolic complications of HSCT include insulin resistance, lipid disorders and hypertension. Both type 1 and type 2 diabetes mellitus are associated with TBI.219The possible mechanisms involved are active areas of investigation.
Declines in neuropsychiatric function following TBI have been observed, but are subtle because of relatively low doses and modern fractionation. A recent prospective study at St. Jude tracked 158 children who had undergone HSCT, of whom 80 had received full-dose fractionated TBI. Patients were given a battery of neurocognitive examinations at 1, 3, and 5 years after transplant. Although TBI was associated with a 3-point decline in IQ compared with non-TBI patients, this difference was small compared with the 20 point difference observed between children from high and low socioeconomic groups.220
Beyond organ-specific toxicities, the risk of developing SMNs is increased in patients that have undergone HSCT. Typically, three groups of SMNs after HSCT are described, and follow a distinct pattern of development (Fig. 15-5):221 MDS and AML, post-transplant lymphoproliferative disorder (PTLD), and solid tumors.222 There are multiple risk factors that may predispose to development of a SMN after HSCT, including but not limited to genetic aberrations, pre-HSCT therapy, conditioning regimen, graft source and processing, post-transplant immunosuppression, and GVHD (for review see DeVita et al.38); only the association of TBI with SMNs is discussed here.
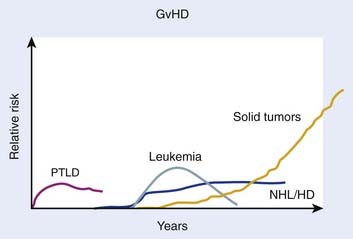
Secondary MDS or AML after HSCT has been reported to occur at rates between 1.1% at 20 months to 24.3% at 43 months. The World Health Organization classifies secondary MDS and AML as alkylating agent– or radiation-related, typically occurring 4 to 7 years after treatment, or topoisomerase-II inhibitor–related, typically occurring 6 months to 5 years after treatment.223 Several studies have attempted to quantify the risk of developing secondary MDS or AML after undergoing TBI, with conflicting results. Studies from research groups from Minnesota,224 Newcastle,225 the City of Hope,226 and France227 have demonstrated no increase in rates of MDS or AML after myeloablative HSCT using TBI. Other studies from Nebraska,228 the European Group for Bone Marrow Transplantation (EBMT),229 Paris,230 Barcelona,231

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


