Topoisomerase 1 inhibitors cure human cancer xenografts in animal models, more so than most other chemotherapy agents. However, their activity in patients with cancer is modest. Ongoing research is studying the optimal analogues that could reproduce animal data in the cancer population. This article analyzes the clinical research with topoisomerase 1 inhibitors in ovarian cancer.
- •
Topoisomerase 1 inhibitors cure human cancer xenografts in animal models, more so than most other chemotherapy agents. However, their activity in patients with cancer is modest.
- •
Ongoing research is studying the optimal analogs that could reproduce animal data in the cancer population.
- •
This article analyzes the clinical research with topoisomerase 1 inhibitors in ovarian cancer.
Topoisomerases relax the DNA supercoiling and perform catalytic functions during replication and transcription. There are two classes of topoisomerases. Type I enzymes cleave one strand of DNA and type II cleave both strands. Six topoisomerase genes have been identified in mammalian somatic cells within these two classes. Type IA enzymes consist of Top3 α and Top3 β; type IB consist of Top1 and Top1mt (mitochondrial); and type IIA consist of Top2 α and Top2 β. CPT is an inhibitor of Top1. Top1 cleaves the DNA phosphodiester backbone, nicking one strand of the DNA duplex and forming a Top1-DNA reversible cleavage complex by covalent bonding of a tyrosine residue. Single-strand breaks induced by Top1 help untangle excessive DNA supercoils during DNA replication and transcription ( Fig. 2 ). Top1 is essential for survival. Mouse embryos lacking Top1 die before the tenth cell division. There are no known wild Top1 mutants. The only mutants detected were in vitro manipulations of cell lines. However, compared with Top1 found in normal tissues, a significant increase of the enzyme was detected in surgical specimens of colon adenocarcinoma; several types of non-Hodgkin’s lymphoma; specimens of leukemia; carcinoma of the stomach, breast, and lung; and malignant melanoma.
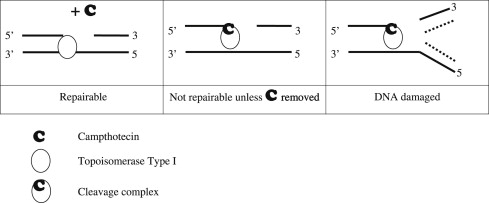
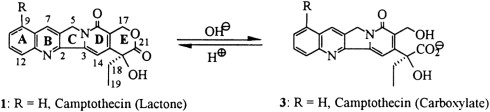
Top1 inhibitors work by stabilizing the Top1 cleavage complexes, leading to DNA damage, because the DNA cannot repair itself. The cytotoxicity of Top1 inhibitors is caused by the paralyzing of Top1 cleavage complexes (where CPT interferes with the Top1/DNA complex by binding to Top1) rather than by the inhibition of Top1 catalytic activity (see Fig. 2 ). Because malignant cells often contain greater amounts of Top1 than normal cells, they should be more sensitive to the toxic effects of the CPT. Binding of CPT to the DNA-topoisomerase complex is reversible and dependent on the chemical structure of CPT. For maximal inhibitory activity, the cleavage complex needs to be maintained. In an acidic milieu, CPT is in lactone form, and in more basic milieu, the equilibrium shifts toward a carboxylate form (see Fig. 1 ). The lactone form is the active form of the drug and the carboxylate form lacks the ability to stabilize the cleavage complex. At the physiologic pH of the human serum, the equilibrium between lactone and carboxylate is in favor of the latter, with the area under the curve (AUC) of the lactone form being below 16%. Additionally, most semisynthetic analogs are poorly water soluble. Two analogs designed to be soluble, irinotecan (CPT-11) and topotecan (NSC 609699), were then synthesized and are currently widely used in clinical practice.
Despite the proved anticancer activity of irinotecan and topotecan, there are major limitations. First, these CPTs are not very active as single agent, with a 10% to 15% response rate in specific cancer indications. They are certainly not curative, despite the potential for inhibiting one of the most crucial enzymes of DNA replication. Both drugs are inactivated within minutes at physiologic pH by lactone ring opening. The E ring–opened carboxylate form has less than 10% potency of the lactone form. Second, the intracellular concentration is reduced by the efflux pumps, although these drugs freely enter the cells by passive diffusion. To overcome these limitations, new CPT derivatives have been developed. In animal models, some of the new analogs are proved more active than irinotecan or topotecan, and a few are even curative of xenograft models instead of simply delaying tumor growth, which is usually the most common observation of anticancer agent activity in animal models. However, when tested in clinical studies none of these newer compounds is better than irinotecan or topotecan. Therefore, we hypothesize that there is either a specific human pharmacodynamics issue that is not understood, or a species response that prevents the replication of animal data in humans. Human blood stabilities of several CPT analogs indicated that serum albumin plays a dual role in the CPT pharmacokinetics. In the case of water-insoluble CPTs, human albumin acts as a sink for carboxylate drug form, binding the carboxylate species and thereby shifting the lactone-carboxylate equilibrium to the right, thus decreasing the potential activity of such CPTs. In the cases of water-soluble CPTs (topotecan, CPT-11), no such preferential binding of the carboxylate drug form by human serum albumin was observed. This might only in part explain the differences seen between human and mouse observed antitumor activity.
Irinotecan
Activity and Metabolism
Irinotecan is chemically unique because of the presence of a bulky side chain on the core five-ring structure, which has to be cleaved for the drug to become pharmacologically active ( Fig. 3 ). The active metabolite of irinotecan is SN-38. As with all CPTs, the carboxylate form is inactive and the lactone ring is the active form, accounting for approximately 60% to 70% of total plasma SN-38 after the end of irinotecan infusion. The conversion of the lactone into the carboxylate form of irinotecan is rapid, with a mean half-life of the lactone form of 9.5 minutes. The volume of distribution of irinotecan at steady state is large, suggesting extensive tissue distribution, and remains unchanged with increasing doses.
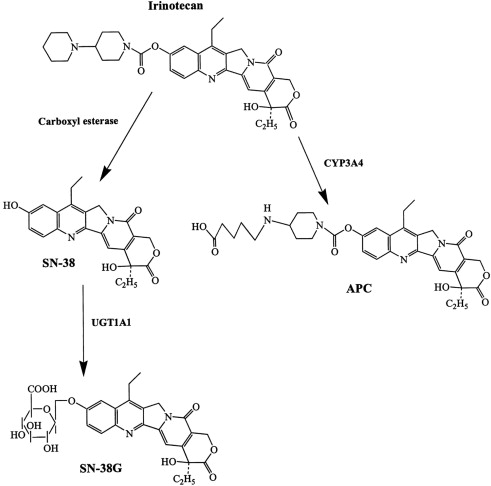
The conversion of irinotecan to SN-38 is an enzyme-catalyzed reaction and liver carboxylesterases (CE; CE-1 and CE-2) are most likely responsible for this conversion. CE-2 plays the most important role in patients with cancer. Because tumors have higher concentrations of CE enzymes compared with normal tissue, the activation of this enzyme may account for the irinotecan activity being higher in specific types of tumors compared with normal tissue. SN-38 itself is further metabolized in human liver by uridine diphosphate glycoronosyltranferase 1A1 (UGT1A1) to an inactive compound, SN-38G (see Fig. 1 ). SN-38G concentrations are proportional to the dose of irinotecan, suggesting that hepatic glucuronidation is not saturated in the dose range studied, up to 600 mg/m 2 . Unlike hepatocytes, other cells in the body have no way of detoxifying SN-38 through glucuronidation, thus contributing to its high toxicity. The activity of UGT1A1 varies among different individuals. For example, in patients with Crigler-Najjar type I syndrome, UGT1A1 activity is totally lacking. The UGT1A1*28 (a 7-TA repeat sequence in the promoter region) genotype is a significant risk factor for severe toxicity by irinotecan, with odds ratio of 7.23. In 2005, the Food and Drug Administration recommended that treatment with irinotecan be altered for individuals with the UGT1A1*28 allele. Because the positive predictive value of the test is only 0.5 and no clear recommendation for dose reduction exists, it is not practical to use UGT1A1 genotyping. A meta-analysis of nine studies including a total of 821 patients revealed a dose relationship between irinotecan and risk of hematologic toxic effects in patients with the UGT1A1 * 28 genotype. Thus, genotype-based decisions should be performed if higher doses of irinotecan ( > 250 mg/m 2 ) are administered, but at low doses the risk of increased toxicity is not defined by this genotype. CYP3A also metabolizes irinotecan into carbonyloxycamptothecin (APC), a metabolite with little cytotoxic activity (see Fig. 3 ). CYP3A pathway is inhibited by many drugs (eg, ketoconazole, loperamide, and ondansetron), thus influencing the concentration of circulating irinotecan. Irinotecan is excreted by the kidney (11%–20%) and the liver (25%–50%). The excretion occurs in the first 24 hours after infusion, with almost complete excretion within 48 hours. When SN-38G is excreted through the bile into the intestines several bacteria, including Escherichia coli , Bacteroides spp, and Clostridium perfringens , can convert the compound back to the active metabolite SN-38 by producing the enzyme β-glucuronidase, potentially leading to late-onset intestinal toxicity (diarrhea). Diarrhea, one of the most common side effects of irinotecan, is associated with SN-38 exposure within the intestinal lumen. Myelosuppression, the second most common side effect of irinotecan, is related to plasma SN-38 exposure.
Administration and Adverse Events
Irinotecan is administered intravenously over 90 minutes with a premedication consisting of atropine 0.25 mg subcutaneously or intravenously to prevent acute cholinergic reactions and a steroid with a serotonin 5-HT3 receptor antagonist to prevent emesis. The patient must be warned about the risk of diarrhea and instructed to take loperamide, 2 mg orally, after each liquid stools. If the diarrhea cannot be controlled within 24 hours, patients must return for intravenous hydration. Prolonged infusion might theoretically improve the efficacy by maintaining inhibition of the cleavage complex, but is not used clinically ( Table 1 ).
| Tumor Type | Indications | Food and Drug Administration Approval/Strength of Recommendation a | References |
|---|---|---|---|
| Metastatic colorectal cancer | First-line therapy, in combination with 5-fluorouracil and folinic acid | Yes | |
| Second-line therapy, single agent after initial 5-fluorouracil–based therapy | Yes | ||
| Carcinoma of esophagus | Nonsurgical | No/Class IIA | |
| Extensive stage small-cell lung cancer | First-line treatment, in combination with cisplatin | No/Class IIA | |
| Acute lymphoid leukemia | Refractory | No/Class IIB | |
| Acute myeloid leukemia | Refractory | No/Class IIB | |
| Carcinoma of cervix | Refractory | No/Class IIB | |
| Extensive stage small-cell lung cancer | First-line treatment, in combination with carboplatin | No/Class IIB | |
| Gastric cancer | Refractory | No/Class IIB | |
| Malignant glioma of brain | Recurrent or progressive disease | No/Class IIB | |
| Metastatic breast cancer | Refractory | No/Class IIB | |
| Non-Hodgkin’s lymphoma | Refractory | No/Class IIB | |
| Non–small cell lung cancer | Refractory | No/Class IIB | |
| Ovarian cancer | Platinum-refractory or platinum-resistant | No/Class IIB | |
| Pancreatic cancer | First-line therapy, in combination with oxaliplatin, 5-fluorouracil, and folinic acid | No/Class IIA |
a Micromedex 2.0. Strength of recommendation definitions. In Edition 2011. Ann Arbor (MI): Thomson Reuters; 2011.
The newest use of irinotecan is for first-line pancreatic cancer in combination with oxaliplatin, 5-fluorouracil, and folinic acid (FOLFIRINOX). In the randomized study of this regimen compared with gemcitabine, the median overall survival of patients with chemotherapy-naive metastatic pancreatic cancer was 11.1 months compared with 6.8 months in the control group (hazard ratio for death, 0.57; 95% confidence interval, 0.45–0.73; P <.001). Median progression-free survival was doubled (hazard ratio for disease progression, 0.47; 95% confidence interval, 0.37–0.59; P <.001), and objective response rate was tripled ( P <.001). The FOLFIRINOX was more toxic, but length of adequate quality of life was doubled.
Topotecan
Topotecan was synthesized in the 1980s as a hydrosoluble semisynthetic derivative of CPT, with a cytotoxic activity similar to the parent compound but a better toxicity profile. Topotecan is a 10-hydroxy camptothecin with a positively charged dimethyl-aminomethyl group at position 9.
Activity and Metabolism
Topotecan, contrarily to irinotecan, is only metabolized to a minor extent. Topotecan undergoes a CYP3A catalyzed metabolism whereby N -desmethyl topotecan, a slightly less active agent, is irreversibly formed. Only low levels of this metabolite have been found in plasma. Both compounds exist in an active lactone and inactive carboxylate form (see Fig. 1 ), but at equilibrium, a minor amount of total topotecan is present in the lactone form. Topotecan and N -desmethyl topotecan may undergo further metabolism to an UGT-mediated glucuronide product, but this transformation is reversible. The kidneys are the most important route of excretion of the drug; hence, renal failure affects the clearance of the drug.
Topotecan has a high volume of distribution, indicating a good tissue penetration. Furthermore, topotecan has a good penetration in cerebrospinal fluid: about 1 hour after the administration, more than 50% of active drug is present in the cerebrospinal fluid. The elimination from cerebrospinal fluid follows a timing curve equivalent to that of elimination from plasma. The fact that the blood–brain barrier is permeable to topotecan led to a series of studies where topotecan was used in patients with brain metastases with or without whole-brain radiation, with short-lasting objective response rates ranging from less than 10% to approximately 50% in patients with various solid tumors.
Topotecan can be used in oral and intravenous formulations, which demonstrated similar efficacy in relapsed small-cell lung cancer.
Administration and Adverse Events
Topotecan is administered as a 30-minute infusion every day for 5 days, every 3 weeks. Unfortunately, this schedule of administration is not very practical, and alternate schedules are being explored, such as 3 consecutive days or weekly in ovarian and recurrent small-cell lung cancer.
The hematopoietic system is the most susceptible organ. Neutropenia, thrombocytopenia, and anemia are the most significant treatment-related toxicity. Gastrointestinal toxicity mainly manifests as nausea. No significant toxic effects have been observed in other organs, such as heart, lungs, liver, kidneys, bladder, and central nervous system ( Table 2 ).
| Tumor Type | Indications | Food and Drug Administration Approval/Strength of Recommendation a | References |
|---|---|---|---|
| Carcinoma of cervix | Stage IVB, recurrent, or persistent, in combination with cisplatin | Yes | |
| Carcinoma of cervix | Advanced, recurrent, or persistent; as monotherapy in patients not amenable to curative treatment with surgery or radiotherapy | No/Class IIB | |
| Ovarian cancer | Metastatic after failure of initial or subsequent chemotherapy | Yes | |
| Small-cell carcinoma of lung | After failure of first-line therapy | Yes | |
| Chronic myelomonocytic leukemia | Refractory | No/Class IIB | |
| Myelodysplastic syndrome | Refractory | No/Class IIB | |
| Nephroblastoma | Refractory | No/Class IIB | |
| Non–small cell lung cancer | Refractory | No/Class IIB |
a Micromedex 2.0. Strength of recommendation definitions. In Edition 2011. Ann Arbor (MI): Thomson Reuters; 2011.
Topotecan
Topotecan was synthesized in the 1980s as a hydrosoluble semisynthetic derivative of CPT, with a cytotoxic activity similar to the parent compound but a better toxicity profile. Topotecan is a 10-hydroxy camptothecin with a positively charged dimethyl-aminomethyl group at position 9.
Activity and Metabolism
Topotecan, contrarily to irinotecan, is only metabolized to a minor extent. Topotecan undergoes a CYP3A catalyzed metabolism whereby N -desmethyl topotecan, a slightly less active agent, is irreversibly formed. Only low levels of this metabolite have been found in plasma. Both compounds exist in an active lactone and inactive carboxylate form (see Fig. 1 ), but at equilibrium, a minor amount of total topotecan is present in the lactone form. Topotecan and N -desmethyl topotecan may undergo further metabolism to an UGT-mediated glucuronide product, but this transformation is reversible. The kidneys are the most important route of excretion of the drug; hence, renal failure affects the clearance of the drug.
Topotecan has a high volume of distribution, indicating a good tissue penetration. Furthermore, topotecan has a good penetration in cerebrospinal fluid: about 1 hour after the administration, more than 50% of active drug is present in the cerebrospinal fluid. The elimination from cerebrospinal fluid follows a timing curve equivalent to that of elimination from plasma. The fact that the blood–brain barrier is permeable to topotecan led to a series of studies where topotecan was used in patients with brain metastases with or without whole-brain radiation, with short-lasting objective response rates ranging from less than 10% to approximately 50% in patients with various solid tumors.
Topotecan can be used in oral and intravenous formulations, which demonstrated similar efficacy in relapsed small-cell lung cancer.
Administration and Adverse Events
Topotecan is administered as a 30-minute infusion every day for 5 days, every 3 weeks. Unfortunately, this schedule of administration is not very practical, and alternate schedules are being explored, such as 3 consecutive days or weekly in ovarian and recurrent small-cell lung cancer.
The hematopoietic system is the most susceptible organ. Neutropenia, thrombocytopenia, and anemia are the most significant treatment-related toxicity. Gastrointestinal toxicity mainly manifests as nausea. No significant toxic effects have been observed in other organs, such as heart, lungs, liver, kidneys, bladder, and central nervous system ( Table 2 ).
| Tumor Type | Indications | Food and Drug Administration Approval/Strength of Recommendation a | References |
|---|---|---|---|
| Carcinoma of cervix | Stage IVB, recurrent, or persistent, in combination with cisplatin | Yes | |
| Carcinoma of cervix | Advanced, recurrent, or persistent; as monotherapy in patients not amenable to curative treatment with surgery or radiotherapy | No/Class IIB | |
| Ovarian cancer | Metastatic after failure of initial or subsequent chemotherapy | Yes | |
| Small-cell carcinoma of lung | After failure of first-line therapy | Yes | |
| Chronic myelomonocytic leukemia | Refractory | No/Class IIB | |
| Myelodysplastic syndrome | Refractory | No/Class IIB | |
| Nephroblastoma | Refractory | No/Class IIB | |
| Non–small cell lung cancer | Refractory | No/Class IIB |
a Micromedex 2.0. Strength of recommendation definitions. In Edition 2011. Ann Arbor (MI): Thomson Reuters; 2011.
Related posts:
 Topoisomerase 1 Inhibitors and Cancer Therapy
Topoisomerase 1 Inhibitors and Cancer Therapy
 Targeting Angiogenesis in Gynecologic Cancers
Targeting Angiogenesis in Gynecologic Cancers
 Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
Role of Histone Deacetylase Inhibitors in the Treatment of Lymphomas and Multiple Myeloma
 mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
mTOR Signaling Pathway and mTOR Inhibitors in Cancer Therapy
 Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
Poly(Adenosine Diphosphate–Ribose) Polymerase Inhibitors in Cancer Treatment
 The Antifolates
The Antifolates
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


