Cancer type
TLRs expressed
Basal cell carcinoma
TLR7
Breast cancer
TLR2, 3, 4, 5, 7, and 9
Brain cancer
TLR2 and 4
Colorectal cancer
TLR2, 3, 4, 5, 7, and 9
Cervical cancer
TLR3, 4, 5, and 9
Esophageal squamous cell carcinoma
TLR3, 4, 7, and 9
Gastric cancer
TLR2, 4, 5, and 9
Human head and neck squamous cell carcinoma
TLR4
Hepatocellular carcinoma
TLR2, 3, 4, 6, and 9
Laryngeal cancer
TLR2, 3, and 4
Lung cancer
TLR2, 3, 4, 7, 8, and 9
Melanoma
TLR2, 3, 4, and 7
Ovarian cancer
TLR2, 3, 4, and 5
Oral squamous cell carcinoma
TLR2 and 4
Pancreatic carcinoma
TLR4 and 7
Prostate cancer
TLR3, 4, and 9
17.2 TLRs Play Important Roles in Human Carcinogenesis
In addition to bacterial and viral components, TLR expression increases in response to inflammation by-products and cellular injury, namely, damage-associated molecular patterns (DAMPs) [13]. Even though TLR7 activation shows antitumor responses in various tumors, including basal cell carcinoma (BCC), breast cancer, and melanoma, it has been postulated that overexpression of TLR7 promotes pancreatic carcinogenesis through mediating several complex pathways [14]. TLR7 is significantly upregulated in both neoplastic ductal epithelial and inflammatory cells, whereas it is undetectable in human normal pancreata. Also, it has been found that TLR7 expression is associated with tumor progression [15]. TLR7 plays important roles in pancreatic carcinogenesis by upregulation of intrapancreatic Notch, MAPK, and NF-κB signaling pathways [15, 16]. It has been discovered that Notch signaling pathway exacerbates inflammation and therefore regulates human pancreatic cancer initiation and maintenance [17]. The NF-κB and MAPK signaling pathways also have proinflammatory effects, mediating TLR7-stimulated pancreatic carcinogenesis [15]. In contrast to TLR7 effects on the pancreas, the expression of TLR4 has been shown to suppress lung carcinogenesis [18], whereas TLR2 expression leads to lung tumor cell progression [19]. Although TLR7 has been considered responsible for intrapancreatic inflammation and fibrosis, destructing exocrine and endocrine organs, its pancreatic carcinogenesis is dependent on baseline levels of inflammation [20]. Moreover, it has been speculated that Kras oncogene is necessary for TLR7-mediated pancreatic carcinogenesis, because no changes have been found in cell cycle regulation and tumor suppressor genes in TLR7-promoted pancreatitis [15]. Collectively, it seems that TLR7-induced pancreatic carcinogenic changes on Kras-transformed cells are secondary to direct effects on peritumoral inflammatory cells, rather than being direct effects of TLR7 stimulation [15].
In addition to TLR7, TLR4 is also involved in colorectal cancer (CRC) tumorigenesis but independent of the presence of baseline inflammation. TLR4 is expressed on CRC cells regardless of the tumor stage [21]. It has been suggested that TLR4 activation is crucial for dysplasia [22]. LPS-stimulated TLR4 activates phosphatidylinositol-3′-kinase (PI3K), leading to phosphorylation of phosphoinositides and, therefore, phosphorylation and activation of Akt. It has been found that PI3K/Akt pathway is expressed in CRC in a stage-dependent fashion [21]. Altogether, TLR7 agonists have been discovered as novel therapeutic approaches for the treatment of BCC and melanoma [23]. However, TLR7 ligation plays opposite roles in pancreatic cancer, indicating the importance of TLR7 signaling blockade in the prevention and treatment of malignancy [18]. Also, targeting of TLR4 signaling pathway in CRC may prevent tumor initiation [12].
17.3 TLR Regulates Tumor-Induced Immune System Response
It has been found that almost all tumor cell lines express single or more commonly multiple TLRs, with TLR4 expression as the highest (Table 17.1). Hsp70 has been found to be highly expressed by tumor cells, playing a ligand role for TLR4. Hsp70-/LPS-mediated TLR4 overexpression leads to the production of nitric oxide (NO) and cytokines such as vascular endothelial growth factor (VEGF), transforming growth factor (TGF), tumor necrosis factor-α (TNF-α), IL-6, and IL-12 p40 [24]. It has been postulated that TLR4 expression is responsible for immune suppression (Fig. 17.1). LPS-stimulated TLR4 expression inhibits T cell proliferation. Also, TLR4-mediated NO suppresses T cell activation [25]. In addition, TLR4-induced IL-6 promotes impairment of Dendritic cells (DCs) maturation, activation of natural killer (NK) T cells, and can also influence NK cell anergy [26]. Furthermore, IL-12 inhibits the generation of allogenic or tumor-specific CTL, contributing to the immune suppression [27].
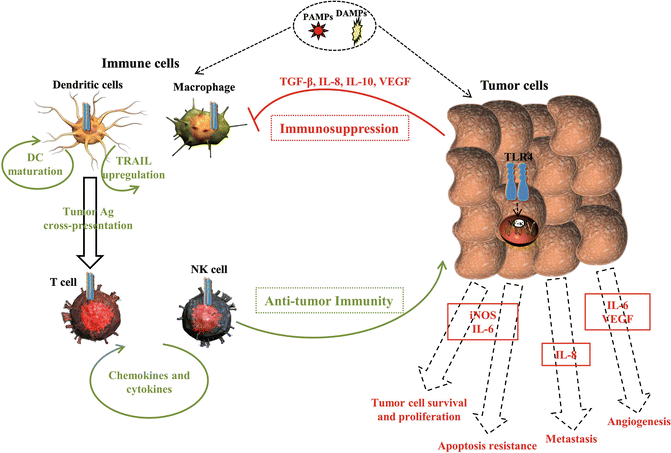
Fig. 17.1
Role of TLR4 signaling in cancer. TLR4 is widely expressed on both immune and tumor cells. TLR4 signaling in cancer is considered a double-edged sword with both pro- and antitumor consequences. TLR4 signaling on immune cells (depicted on the left-hand side in green color) enhances antitumor immunity by cytokine/chemokine upregulation, DC maturation, and function. TLR4 is also responsible for efficient tumor antigen cross-presentation. Alternatively, TLR4 signaling on tumor cells (depicted on the right-hand side in red color) increases their tumorigenic activity
On the other hand, upregulated TLR4 increases B7-H1, B7-H2, and CD40 levels but decreases Fas expression on tumor cells, thereby leading to cancer cell escape from immune system surveillance and CTL attacks [24]. Therefore, TLR4 plays an important role in the protection of tumor cells from the immune system response (Fig. 17.1); nonetheless, it has been suggested that TLR4 function is necessary for DC maturation and CD4+ CD24+ regulatory T cell blockage [28].
TLR4 is highly expressed in both cell membrane and cytoplasm of human oral squamous cell carcinoma (OSCC) [29]. The expression is associated with tumor cell differentiation, and TLR4 level is significantly higher on well- and moderately differentiated tumor cells when compared to poorly differentiated cancer cells. LPS-stimulated TLR4 activates both NF-κB and p38 MAPK pathways, leading to the massive production of IL-6, IL-8, and VEGF. IL-6 is considered as a principle biomarker of poor prognosis in several human cancers [29]. Higher levels of IL-6 can lead to tumor progression, resistance to apoptosis, chemoresistance [30], tumor angiogenesis, and tumor invasion [31]. IL-8 plays anti-apoptotic roles and promotes tumor metastasis [32]. VEGF is involved in angiogenesis and immunosuppression and also suppresses DC number and differentiations [33]. These results indicate the crucial effects of TLR4 signaling in human OSCC survival and metastasis, therefore suggesting the importance of novel approaches targeting TLR4 signaling pathway for OSCC treatment.
Although TLR2, TLR3, and TLR4 are expressed in normal primary melanocytes, they are significantly overexpressed on most melanoma cell lines [34]. The presence of TLRs on normal melanocytes plays important roles in the recruitment of innate immune cells. Overexpression of TLRs in melanocytes leads to chronic inflammation, thereby increasing the risk of tumor development and progression [35]. Upregulated TLR2, TLR3, and TLR4 promote production of proinflammatory cytokines (TNF-α, IL-1, IL-6, and granulocyte colony-stimulating factor (GCSF)) and chemokines (CCL2 and CXCL10). Also, these TLRs stimulate the secretion of IL-10 and cyclooxygenase-2 (COX-2) (inflammatory factor) [34]. Higher levels of TNF-α induces IL-6 and CCL2 synthesis, leading to the tumor progression. Also, TNF-α regulates infiltration of leukocytes in cancers by chemokine modulation [36]. Besides, CCL2 and CXCL10 promote escalating inflammation and immunity in melanoma cancer [37]. Additionally, TLR3 triggers NF-κB-mediated upregulation of inflammatory molecules and recruits leukocytes, promoting anticancer immune responses [38]. TLR4 is found to be highly expressed in breast cancer cells. It has been found that targeting of TLR4 signaling by TLR4AsiRNA leads to significant inhibition of breast cancer cell proliferation. Also, inhibition of TLR4 interrupts its downstream signaling pathway, leading to the strikingly depressed levels of IL-6 and IL-8, and, therefore, attenuates tumor cell survival by decreasing their resistance to cytotoxic T lymphocyte (CTL) and natural killer cell (NKC) attack. These results suggest that targeting of TLR4-mediated signaling pathway by TLR4AsiRNA is a novel promising strategy for breast cancer treatment, although this inhibition may promote other cancers, including lung cancer [39]. Thus, manipulation of TLR4 should be done with precise attention considering its possible interactions. Additionally, LPS-stimulated TLR4 upregulation promotes NF-κB signaling pathway and contributes in the production of inflammatory cytokines (including IL-6 and IL-8), VEGF, and granulocyte-macrophage colony-stimulating factor (GM-CSF), leading to tumor progression and development of myeloid-derived suppressor cell (MDSC) [40]. MDSC can promote chronic inflammation and also immune suppression by stimulation of regulatory T cell function [41].
On the other hand, flagellin-stimulated TLR5 leads to the production of various chemokines such as epithelial cell-derived neutrophil-activating peptide-78 (ENA-78), macrophage inflammatory protein 3α (MIP3α), monocyte chemotactic protein-1(MCP-1), macrophage-derived chemokine (MDC), IL-6, Gro-α, and osteoprotegerin, which are involved in monocyte, leukocyte, and neutrophil attraction [42]. TLR5-induced infiltration of immune cells, including neutrophils, suppress proliferation marker PCNA, promoting strong antitumor response through tumor necrosis and inhibition of tumor growth [42]. Thus, flagellin-induced TLR5 expression can be used as a novel therapeutic approach for human breast cancer.
17.4 TLR Targeting May Inhibit Cancer Cell Proliferation
TLR7 expression suppresses phosphatase and tensin homologue deleted on chromosome 10 (PTEN) [15]. Suppressed levels of PTEN lead to PI3K/Akt pathway activation and increased level of TGF-β, mediating phosphorylation and activation of STAT3 [43]. STAT3 acts as a proinflammatory marker and central to neoplastic progression in pancreatic tumor [44]. TGF-β promotes cancer invasion [45], and PI3K/Akt signaling pathway stimulates tumor cell proliferation, thus leading to tumor progression [46]. Also, it has been suggested that TLR4 has proproliferative roles. It has been found that human head and neck squamous cell carcinoma (HNSCC) expresses almost all TLRs for its own benefit. TLR4 has been shown to be highly expressed in well- and moderately differentiated HNSCC but weakly present on poorly differentiated cells [40]. It has been suggested that well-differentiated cells contain higher amounts of bacteria and bacterial products, thereby leading to higher expression of TLR4. LPS-induced expression of TLR4 can phosphorylate Akt, thus increasing tumor cell proliferation [40].
17.5 TLR Triggering Can Promote Antitumor Response
It has been reported that TLR5 is overexpressed in gastric cancer cell, leading to strong antitumor immune response and suppression of tumor growth [47]. In contrast, early activation of TLR5 has been shown to promote tumor growth in mouse mammary cells. High levels of TLR5 have been found in invasive ductal carcinoma cells, whereas moderate expression is observed in medullary carcinoma and invasive lobular carcinoma. Flagellin-induced expression of TLR5 in breast cancer cells increases phosphorylation of IκB, ERK, JNK, STAT1, and STAT3, leading to the induction of inflammatory cytokines (such as TNF-α, IL-1β, IL-6, and IL-8) mRNA. This flagellin-stimulated cytokine production leads to decreased level of proteins contributed in the cell cycle and inversely increased level of CDK inhibitor 27, thereby inhibiting breast cancer cells proliferation and colony formation. However, it has been found that flagellin fails to induce cancer cell apoptosis [42].
17.6 Regulatory Effects of TLRs on PI3K/Akt Signaling Controlling Tumor Progression
Akt has been known to promote cyclinD1 and c-Myc expression by targeting the kinase PI3K/Akt mammalian target of rapamycin (mTOR), which leads to proliferation of various cancer cells [48]. Also, Akt inhibits GSK-3b phosphorylation and therefore suppresses β-catenin nuclear translocation [49]. In addition, Akt regulates cell death through decreasing levels of pro-apoptotic molecules, such as caspase-9, p53, NOXA, and PUMA [50]; however, it inversely regulates increasing anti-apoptotic molecule levels including XIAP, Bcl-xL, and Mcl-1 [51]. Moreover, Akt functionally suppresses both p21Wsf1/Cip1 and p27Kip1 that are negative regulators of the cell cycle [52]. Furthermore, the presence of phosphorylated Akt has been reported to be associated with advanced stages of tumor and poor clinical prognosis [53].
Several TLRs have been detected on human prostate cancer cells. TLR3 and its ligand polyinosinic-polycytidylic (poly(I:C)) acid negatively regulate Akt-mediated pathways in human prostate cancer cells. Poly(I:C) dephosphorylates Akt and therefore impairs PI3K/Akt pathway, leading to the inhibition of cell proliferation by downregulation of cyclin D1 and c-Myc and upregulation of p21Wsf1/Cip1 and p27Kip1 [54]. Also, poly(I:C) increases β-catenin translocation into the nucleus [49, 54]. The PI3K/Akt pathway has also been found to play potent roles in CRC progression and metastasis. TLR4 is responsible for the activation of PI3K/Akt pathway and therefore promotion of tumor progression. Moreover, it has been reported that TLR4 targeting can prevent liver metastasis and burden of the tumor [55]. However, TLR4 pathway targeting seems to be a novel valuable therapeutic approach for the prevention of CRC progression and metastasis.
17.7 TLR-Mediated Hypoxia-Inducible Factor 1 (HIF-1) Expression Leads to Tumor Progression
It has been found that HIF-1 is involved in tumor progression [12]. In hypoxic conditions, HIF-1α stabilizes and binds HIF-1β, leading to the active form of HIF-1 [56], but, in normoxic situations, oxygen-sensing prolyl hydroxylases degrade HIF-1α and keep its level low [57]. Poly(I:C)-induced TLR3 increases the specific I.3 isoform of HIF-1α expression and HIF-1 complex nuclear accumulation in normoxic environment. TLR3’s effect on the enhancement of HIF-1α expression is based on the increase of HIF-1α translation rather than prevention of its degradation [58]. Higher levels of HIF-1α have been detected in prostate cancer bone metastasis indicating the importance of HIF-1α in prostate tumor prognosis [59]. It has been reported that poly(I:C)-stimulated TLR3 leads to the upregulation and nuclear translocation of HIF-1α in more advanced prostate cancer cells. Overexpressed HIF-1 increases VEGF secretion [12]. VEGF promotes neovascularization in hypoxic tumor space, leading to tumor progression [60]. HIF-1α complex upregulates anti-apoptotic genes including Bcl-xL, survivin, and MCL-1 [61]. Moreover, the complex impairs caspase-3 function, inhibiting TLR3-mediated apoptosis of progressed prostate cancer cells. However, forcing the upregulation of the HIF-1α-isoform 3 in less aggressive prostate cancer cells can lead to HIF-1 complex nuclear accumulation secondary to the poly(I:C) stimulation. It seems that differential expression levels of HIF-1α in different stages of prostate cancer cells regulate the tumor cell’s response to TLR3 stimulation [12]. However, HIF-1α level should be precisely regulated through changes in TLR signaling pathway.
17.8 Role of TLRs in Tumor Cell Lysis and Apoptosis
TLR3 and TLR7 have been found to be effective in increasing γδ T cell cytotoxicity and cytokine production [62]. It has been reported that γδ T cells play important roles in tumor cell lysis by massive production of IFN-γ and TNF-α. Also, γδ T cells secrete perforin, granzymes, and TNF-α apoptosis-stimulator ligands, mediating tumor cell lysis [63]. The cytotoxic effect of γδ T cells increases in response to poly(I:C)-stimulated TLR3 overexpression. Additionally, γδ T cell-secreted cytotoxic mediator levels increase in tumor cells secondary to poly(I:C)-induced TLR3 overexpression and imiquimod-stimulated TLR7 upregulation. In the presence of γδ T cells, poly(I:C)-mediated TLR3 activates NF-κB p65 and caspase signaling, leading to IFN-β production and apoptosis [64]. Imiquimod-induced TLR7 also increases MyD88 and NF-κB signaling pathways, leading to caspase pathway activation and therefore resulting in tumor cell death [62].
It has been reported that the activation of killer receptor NKG2D, which binds to the stress-inducible MHC class I chain-related antigens (MIC) A/B and UL16-binding proteins (ULBP) 1–4, is crucial for the cytotoxic activity of γδ T cells [65]. Poly(I:C)-stimulated TLR3 leads to the production of TNF-α and, therefore, CD54 expression [66]. Although imiquimod-induced TLR7 decreases MHC class I molecules on tumor cells, imiquimod fails to increase CD54 levels. The presence of CD54 and NKG2D may increase the ability of γδ T cell-mediated tumor lysis. These results indicate that several pathways are involved in tumor cell lysis [62]; nevertheless, it seems that TLR3 and TLR7 are involved in the cytotoxic function of γδ T cells, and proper regulation of these TLRs may bring new treatment hopes for cancer patients. TLR7 activation also leads to the induction of STAT3, which occurs simultaneously with increasing proliferative and anti-apoptotic genes such as c-Myc and Bcl-xL [15]. It has been reported that a high c-Myc level acts as a prognostic factor in advanced pancreatic tumor, and also its level is associated with poor survival in patients suffering from pancreatic cancer [46]. On the other hand, TLR7 upregulation impairs G1 phase control by downregulation of cyclin D1 and also increasing cyclin B1, leading to the G2 to M phase transition [15].
It has been suggested that tumor cell’s resistance to the drug-induced apoptosis originates from TLR4-mediated Akt phosphorylation. On the other hand, it is reported that TLR4 leads to the translocation and binding of p65 subunit of NF-κB to DNA, thereby leading to the inhibition of cisplatin-induced apoptosis and NK cell-mediated tumor lysis. Also, TLR4-activated NF-κB, MyD88, and IRAK4 are associated with tumor progression, as these factors play anti-apoptotic and inflammatory roles. In addition, TLR4 has been considered responsible for tumor cell resistance to chemotherapy, suggesting TLR4 pathway targeting as an important novel treatment strategy for HNSCC [40]. During the targeting of the TLR4 signaling pathway, beneficial effects of TLR4 stimulation should be harnessed while eliminating the possible negative ones (Fig. 17.1). Therefore, it has been speculated that TLRs work like a double-edged sword, stimulating host immune reaction against tumor on one hand and promoting tumor progression on the other.
Moreover, poly(I:C)-induced expression of TLR3 promotes cancer cell apoptosis by caspase upregulation, with the induction of p53 and its pro-apoptotic target NOXA. In addition to apoptosis induction by poly(I:C), the ligand can induce autophagy that is cytoprotective toward apoptosis, indicating the inverse association of apoptosis and autophagy [54].
17.9 TLRs are Involved in Tumor Metastasis
It has been accepted that the upregulated expression of TLR3 leads to increased chemokine (C–C motif) ligand 5 (CCL5) and IL-6 levels. It has been suggested that cancer cell migration and perineural invasion is mediated by TLR3-induced CCL5 and IL-6 [9]. CCL5 increases matrix metalloproteinase 9 (MMP-9) and, therefore, inhibits T cell antitumor response, leading to angiogenesis and tumor growth [67]. On the other hand, activated NF-κB stimulates genes that are involved in cell differentiation, cell invasion, and anti-apoptotic protein production, such as HIF-1α [12] and apoptotic protein-2 inhibitor [68]. It has been speculated that higher levels of TLR3 in breast malignancy and HNC is strongly associated with tumor invasion and metastasis [69]. The administration of bafilomycin A1 (BA1) which antagonizes TLR3 leads to decreased levels of CCL5 and IL-6, therefore controlling tumor aggressive behavior [69]. Also, TLR4 activation has been found to be responsible for apoptosis resistance in ovarian cancer cell [70]. These results highlight the importance of TLR targeting in the prevention of tumor progression and metastasis. Furthermore, upregulation of COX-2 has been found to be associated with an aggressive type of melanoma cancer. Interestingly, Goto et al. have found that TLR-mediated signaling pathway (MyD88 and NF-κB) is also responsible for melanoma tumor cell migration [34]. These results show that TLRs play principal roles in the progression of melanoma cells, thereby suggesting the beneficial effect of targeting TLR signaling pathways in discovering a novel therapeutic approach for melanoma. It has been reported that TLRs are also involved in cancer recurrence and metastasis [55]. Tumor resection is a choice treatment; however, 30 % of patients with grade III CRC and 10 % of patients with grade I/II suffer from recurrence 5 years after curative surgery [71]. It has been found that surgical resection can induce local recurrence or distant metastasis [72]. Recently, it has been suggested that systemic inflammation and postoperative infection are associated with CRC recurrence [73]. TLR4 has been found to be highly expressed in patients with liver metastasis and poor clinical outcome [74]. Upon infection, LPS-induced upregulation of TLR4 leads to physical interaction of PI3K with MyD88, leading to phosphorylation of Akt and, therefore, β1 integrin activation, which is the main subunit for collagen binding. LPS-stimulated TLR4 and β1 integrin are responsible for endothelial adhesion by enhancing cancer cell’s binding mostly to type I/IV collagen and less to fibronectin and laminin [75]. Additionally, TLR4-mediated signaling promotes hepatic involvement and liver metastasis [76]. Although few studies have found that TLR4-induced cascade plays proliferative and anti-apoptotic roles in cancer cells, leading to cancer metastasis [77], the same results were not obtained in other studies [55]. This LPS-induced signaling suggests a novel therapeutic target for preventing recurrence or metastasis in patients who were treated by curative resection of colorectal cancer. Three targeting approaches such as TLR4 targeting by eritoran, PI3K inhibition by PI 103, and β1 integrin functional blockage by anti-β1 integrin antibody have been suggested. Since PI3K and β1 integrin play important roles in several normal processes and also LPS-induced TLR4 signaling-mediated events in cancer cells, TLR4 targeting strategy seems to be a better therapeutic approach in patients with CRC [52]. Thus, targeting of TLR4 signaling pathway can be beneficial for patients both with and without postoperative infection.
Related posts:
 Novel Strategy of Cancer Immunotherapy: Spiraling Up
Novel Strategy of Cancer Immunotherapy: Spiraling Up
 Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
Dendritic Cell Vaccines for Cancer Therapy: Fundamentals and Clinical Trials
Psychoneuroendocrinoimmunotherapy of Cancer
 Polarization of Tumor Milieu: Therapeutic Implications
Polarization of Tumor Milieu: Therapeutic Implications
 Photodynamic Therapy and Antitumor Immune Response
Photodynamic Therapy and Antitumor Immune Response
 Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Role of γδ T Lymphocytes in Cancer Immunosurveillance and Immunotherapy
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


