Fig. 8.1
Synergistically increased risk for HCV-mediated HCC by alcohol abuse or/and obesity. Comorbidity such as alcohol abuse and obesity synergistically heightens the risk of developing HCC by 6- to 8-fold in HCV-infected patients. As such HCV-infected, alcoholic or obese patients have 40–60 times higher HCC risk compared to healthy subjects
The most challenging aspect of HCC treatment is its refractoriness to chemotherapy. Among many potential mechanisms which underlie chemoresistance, the role of tumor-initiating stem cell-like cells (TICs) or the so-called cancer stem cells (CSCs) has received a spotlight. Stem cells have three major characteristics, self-renewal, asymmetric division (clonality), and plasticity. Forty percent of HCC are assumed to have clonality and to originate from progenitor/stem cells [13–16]. CD133+/CD49f+ cells in liver tumors correlate with tumorigenesity and the expression of “stemness” genes, such as Wnt/β-catenin, Notch, Hedgehog/SMO, and Oct3/4 [17–19]. Indeed, CD133+/CD49f+ HCC CSCs are chemoresistant [20] and survive during an initial therapy. Although an encouraging therapeutic response may be seen, survived CSCs eventually establish a clonal expansion and tumor recurrence. This chemoresistance may be caused by the plasticity of CSCs with dysregulated signaling and gene expression. Several oncogenic signaling pathways are activated in HCC or CSCs, including PI3K/AKT [21], signal transducer and activator of transcription 3 (STAT3) [22, 23], and hedgehog [24, 25] while defective tumor suppressor transforming growth factor-beta (TGF-β) pathway is also implicated [26, 27]. Another pivotal mechanism is asymmetric division of CSCs producing dormant daughter cells which are less sensitive to chemotherapeutic drugs.
8.2 Ectopic TLR4 Activation Underlies HCV-Alcohol Synergism
In our efforts to establish mouse models of HCV-alcohol synergism for HCC, we fed first alcohol to HCV Ns5a transgenic mice by using the mouse intragastric feeding (iG) model. This approach is prompted by the observation that TLR4 expression is induced in the liver by hepatocyte-specific NS5A expression [28]. As endotoxin-TLR4 pathway is established in the pathogenesis of ALD [29, 30], a synergism between alcohol-mediated endotoxemia and NS5A-induced TLR4 overexpression was predicted. Indeed, alcohol feeding to the Ns5a mice results in aggravated liver damage with severe hepatitis induced in some mice [31]. This pathology is dependent on TLR4 as it is abrogated in alcohol-fed Ns5a:Tlr4−/− mice. This synergism is also due to endotoxin as it is attenuated by the concomitant treatment of the mice with polymyxin B and neomycin and is conversely potentiated by intragastric administration of LPS. To extend this observation to an extended period of 12 months, a modified ethanol-containing liquid diet was fed to Ns5a and Ns5a:Tlr4−/− mice. Although no liver tumor is observed in none of wild type (WT) or Ns5a:Tlr4−/− mice fed alcohol, 23 % of alcohol-fed Ns5a Tg mice developed liver tumors [31]. This TLR4-dependent tumorigenic phenotype was subsequently reproduced in alcohol-fed HCV Core Tg mice [32].
8.3 Identification of TLR4/NANOG-Dependent TICs
Immunostaining of liver tumor sections from alcohol-fed Ns5a mice revealed cells double-positive for NANOG and CD133 or CD49f [31]. This prompted a FACS analysis of cells dissociated from liver tumors of these mice which detected a small yet significantly increased percentage of CD133+/CD49f+ cells as compared to WT mice (1.11 % vs. 0.05 %). Gene profiling analysis of sorted CD133+/CD49+ cells shows consistently upregulated stemness genes such as Nanog, Oct4, Sox2 as compared to CD133−/CD49+ or CD133−/CD49f− cells.
This heightened expression of stemness genes and cell proliferation are largely reduced by TLR4 silencing with lentiviral short-hairpin RNA (shRNA) compared to control CD133−/CD49f+ cells [32]. The CD133+/CD49f+ cells form colonies in soft agar and spheroids in ultra-low-adhesion plates, demonstrating they have anchorage-independent growth and self-renewal ability [32]. Subcutaneous transplantation of CD133+/CD49f+ cells, but not CD133−/CD49f+ cells into immunocompromised (NOG) mice, following infection with a red-fluorescence (dsRed) lentiviral vector, results in tumor development; and this tumor growth, assessed by dsRed imaging, is inhibited by Tlr4 or Nanog silencing by lentiviral shRNA transduced prior to transplantation [32] (Fig. 8.2), indicating that CD133+/CD49f+ cells are TLR4/NANOG-dependent TICs and that Tlr4 is a putative proto-oncogene involved in the genesis/maintenance of TICs and liver tumor in HCV Tg models. Furthermore, these NOG mice derived tumor have tumor-initiation capacity since injection of serial transplantation of these tumors into another NOG mice generates tumors in NOG mice. Then one will raise a question where these TICs are generated. These TICs may have been generated from hepatoblasts in several etiologies since hepatoblastic HCC subtype with poor prognosis has a gene expression profile with markers of hepatic oval cells, suggesting that this subtype of HCC arises from LPCs [33–36]. Indeed, these HCC often recurs after chemotherapy presumably due to the presence of chemo-resistant TICs [37]. These aspects need further investigation.
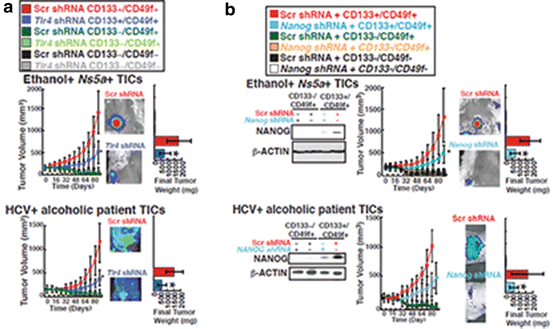
Fig. 8.2
TICs’ tumorigenic activity is dependent on TLR4 and NANOG. CD133+/CD49f+ TICs isolated from liver tumors of alcohol-fed Ns5a Tg mice or alcoholic HCV patients were transplanted subcutaneously into NOG mice following lentiviral transduction of EGFP to allow a whole animal imaging for assessment of tumor growth for a period of 80 days. To test the dependence on TLR4 and NANOG, they were respectively knocked down by lentiviral expression of specific or control shRNA prior to transplantation. Tumor volume was calculated by three dimensional assessment of the tumor image, and final tumor weights were also compared at the end of the experiment. Note TLR4 (a) or NANOG (b) knockdown significantly attenuates tumor growth derived from mouse and patient TICs. Immunoblotting of cell lysates collected 10 days after the transplantation confirms expression of NANOG in the TICs and knockdown of this protein with specific shRNA. *p < 0.05 (Reproduced from the published figure of the reference [32] with permission)
8.4 TLR4 as a Putative Proto-oncogene
The obligatory roles of endotoxin and TLR4 in HCC promotion are shown in various etiological settings including initiation in alcohol-HCV model and promotion [32] in the DEN/CCl4 model [38]. Activated TLR4 pathway is responsible for promotion of HCCs from different animal models [38], which offer new therapeutic targets for HCC. As PAMP (pathogen-associated molecular pattern), including TLR4, mediates inflammatory responses to endotoxin and other ligands, inflammation is strongly associated with cancer in other cancers, including lung [39], colon [40], and skin carcinomas [41]. While TLR expression is very heightened in macrophages and lymphocytes, normal hepatocytes have less or nonfunctional TLR4, but ectopically expressed-TLR4 in epithelial cells is involved in oncogenesis as studies in other cancer models implicate liver has also similar oncogenic pathways since gut-derived endotoxin directly damages liver due to proximal anatomy of gut and liver through portal veins [31, 38, 42]. As we mentioned above, we have shown that long-term (12 months) feeding of alcohol diet induces liver tumors in HCV Ns5a Tg mice [43–46] and these incidences are reduced if the mice were crossbred with defective Tlr4 mice [32]. Plasma LPS levels are elevated equally in both TLR4 sufficient and deficient mice fed these diets, indicating that ligand level is not changed even by disruption of its receptor Tlr4 [31, 32]. Indeed, tumor tissues from Tlr4+/+ models display accentuated expression of TLR4 and its activation as assessed by its downstream marker TRAF6-TAK1 complex formation [32]. Furthermore, activation of human TLR4 oncogenic pathway, especially NANOG overexpression, is also noted in HCC sections of alcoholic HCV as well as non-steatohepatitis (NASH) patients [32], supporting this activation of TLR4-NANOG axis is clinically relevant for the development of both human and mouse HCCs [31]. TICs are resistant to chemotherapy and are associated with metastatic HCC, which is commonly observed in HCV-infected patients with alcohol abuse. Sensitization of TICs to chemotherapy and identifying therapeutic molecular targets could be a considerable savings in morbidity, mortality, and cost. However, in HCCs, there are many signaling involved in genesis of HCCs. In the next section, one of the typical crosstalk between TLR4 and TGF-β pathways will be discussed.
8.5 TLR4 and TGF-β Mutual Antagonism in Liver Tumorigenesis
Our study identifies TLR4 signaling as a central mediator in synergistic liver tumor formation by HCV and alcohol via the genesis of TLR4/NANOG-dependent TICs. On the other hand, deficient TGF-β pathway caused by inactivation of at least one of the TGF-β signaling components is a well-known risk factor for HCC in man [26, 47] and a causal oncogenic mechanism in animal models [15, 48]. Thus, we wondered about the relationship between the two pathways. To investigate this question, we have used two complementary approaches. First, we looked for TIC-specific genes which may be involved in regulating TGF-β pathway by screening lentiviral cDNA library established from TIC vs. CD133−/CD49f+ control cells for transformation of the p53 deficient hepatoblast cell line PL4 [32]. This has identified Yap1 and Igf2bp3 as two TIC-associated genes which are under direct transcriptional control of NANOG and contribute to TICs’ tumor-initiating activities both in vitro and in vivo [32]. Further, these two gene products are shown to inhibit the TGF-β tumor suppressor pathway at the two distinct levels but in an interactive manner. YAP1 associates with SMAD3 and SMAD7 to block nuclear translocation of p-Smad3. IGF2BP3, an mRNA binding protein, promotes IGF-II translation by binding to the 5′ UTR of Igf-II mRNA [49]. IGF-II activates AKT and subsequently mTOR, which suppresses SMAD3 activation [50]. Indeed, mTOR activation by IGF2BP3 inhibits phosphor-activation of SMAD3 as such Rapamycin increases p-SMAD3 in TICs or even abrogates suppressed p-SMAD3 level caused by a constitutively active AKT. This IGF2BP3-AKT-mTOR pathway also interfaces with the YAP1-SMAD3/SMAD7 pathway described above. Activated AKT phosphorylates Ser-127 of YAP1, and p-Ser127-YAP1 interacts most actively with p-SMAD3 for cytosolic SMAD3 retention. Thus AKT activated by IGF2BP3 facilitates dual actions of mTOR-mediated suppression of SMAD3 phosphor-activation and p-YAP1-mediated p-SMAD3 retention, resulting in effective blockade of TGF-β pathway. As expected, silencing of Igf2bp3 and Yap1 in TICs restores TGF-β pathway with increased nuclear p-SMAD3, reduces TICs’ tumorigenic activity, and enhances the chemosensitivity of TICs in vivo [32].
We have also used a reverse approach to test the reciprocal TLR4-TGF-β antagonism by assessing TLR4 expression and activation in Spnb2+/– mice. In fact, it is well known in innate immunity that the lack of a functional TβRII [51, 52] or Smad3 [53] results in extensive inflammation due to increased TLR4 expression and LPS hyper-responsiveness [54]. We believe this reciprocal regulation of augmented TLR4 response due to deficient TGF-β signaling also plays a critical role in generation and oncogenic activity of Nanog+ CSCs. SPNB2 is the chaperone protein which recruits p-SMAD3/SMAD4 into the nucleus, and SPNB2 haploinsufficiency in Spnb2+/– mice reduces TGF-β signaling and causes spontaneous development of HCC [15]. TLR4 expression is induced in the liver of this genetic mouse model as compared with WT mice. Feeding Spnb2+/– mice with alcohol for 12 months results in conspicuous TLR4 activation as assessed by TAK1/TRAF6 interaction and doubles the liver tumor incidence as compared to Spnb2+/– mice fed with a control diet [32]. More importantly, this increment of the tumor incidence is completely abrogated in alcohol-fed Spnb2+/-Tlr4−/− compound mice, demonstrating reciprocally upregulated TLR4 in Spnb2+/– mice with reduced TGF-β signaling, is also responsible for alcohol-associated liver tumorigenesis in the model. We readily extend this concept to clinically more relevant cells, the Huh7 human HCC cell line. Knockdown of SPNB2 in these cells increases TLR4 expression and tumorigenic activity in NOG mice [32].
8.6 Anabolic Metabolism and TIC Self-Renewal
A critical event leading to deregulated TIC proliferation is inactivation of the p53 tumor suppressor [55, 56], which acts as a key barrier against pluripotency and stem cell proliferation. This function of p53 is carried out through direct repression of stemness-associated transcription factor (TF) network components [57]. Mutation or loss of p53 is found recurrently in diverse malignancies including HCC [58] and is associated with poor prognosis [59, 60]. Strikingly, genetic inactivation of p53 also leads to loss of cell polarity and aberrant execution of self-renewal [61–63]. The cell polarity determinant and tumor suppressor NUMB stably interacts directly with p53, protecting it from ubiquitin-mediated proteolysis catalyzed by the MDM2 E3 ubiquitin ligase [64]. In polarized epithelial cells and in untransformed progenitor cells, NUMB is distributed asymmetrically and segregates into the daughter cell that proceeds to differentiate. Cells deficient in p53 fail to correctly localize NUMB and lose this intrinsic polarity [65, 66], however little is understood about the composition and regulation of the Numb-p53 complex.
We examined biochemically the composition of the Numb-p53 complex in CD133+/CD49f+ TICs isolated from liver tumors of alcohol-fed HCV Ns5aTg mice, Fractionation of TIC lysates using sucrose density gradient centrifugation revealed that NUMB and p53 are the constituents of a high molecular mass (>700 kDa) complex, which is disintegrated upon NANOG-mediated activation of aPKCζ, a NUMB kinase [67]. Using affinity purification and tandem mass spectrometry, we identified the ATG8/LC3-binding protein TBC1D15 as a novel component of this high molecular mass complex. Enforced expression of TBC1D15 reduces steady state levels of p53 and this effect is blocked by a Nutlin-3 treatment, suggesting that TBC1D15 triggers the MDM2-dependent degradation of p53.
TBC1D15 is comprised of two distinct structural domains: a C-terminal GTPase activating protein (GAP) domain that inactivates the Rab7 GTPase, which mediates endosome/autophagosome-fusion to lysosomes [68, 69] and a functionally uncharacterized N-terminal domain. We expressed Flag-tagged variants of each domain individually with myc-tagged p53 (myc-p53), and found that the N-terminal domain (Flag-TBC1D15-N) recapitulated inhibition of myc-p53. Destabilization of myc-p53 corresponded closely with the extent of its displacement from NUMB. Sequence analysis of the N-terminal domain revealed a 50 amino acid region containing significant homology to the Drosophila protein CANOE, which regulates the localization of cell-fate determinants during asymmetric division and interacts genetically with numb [70, 71]. Coexpression of myc-p53 with GFP fusion proteins containing either the CANOE homology region (TBC-cno, aa 159–270) or the N-terminal region (TBC-N1, aa 2–158) revealed that GFP-TBC-N1 but not GFP-TBC-cno associated stably with NUMB, suggesting that a primary sequence or higher order structural motif within this region of TBC1D15 directly binds to NUMB and dissociates it from p53 to promote p53 degradation. However, the mutual requirements and relative contributions of TBC1D15 and aPKCζ-mediated NUMB phosphorylation for p53 dissociation and degradation are not yet fully understood and merit further investigation.
We also found that Tbc1d15 is transcriptionally repressed by p53, revealing a mutually antagonistic regulation between these genes. In agreement with these findings, three human HCC cell lines express TBC1D15 at higher levels than primary hepatocytes. In particular, Hep3B cells with p53 deficiency and Huh7 cells with mutant p53 express substantially higher TBC1D15 than HepG2 cells which have wild type p53. Thus, p53 levels are inversely correlated with TBC1D15 expression in these cells. Similarly, TBC1D15 levels are increased in TICs compared to CD133− cells, and TBC1D15 is strongly expressed in tumors arising from TICs implanted subcutaneously into mice, as determined by immunohistochemical analysis of sectioned tumors.
Interestingly, the TCTP oncoprotein was also found in association with the Numb-p53 complex and shown to stimulate MDM2-mediated proteolysis of p53 [72]. These results, along with our recent data on TBC1D15, collectively suggest that the Numb-p53 complex may serve as a pivotal control platform that integrates multiple inputs to permit the rapid modulation of cellular p53 levels. As there appears to be no significant primary sequence homology between TCTP and TBC1D15, these proteins may dock with distinct subunits in the NUMB-p53 complex.
Cellular levels of TBC1D15 are diminished through starvation-induced autophagic degradation, triggered through acute nutrient deprivation, depletion of ATP or chemical inactivation of the mTOR kinase complex. Conversely, TBC1D15 accumulates when autophagic flux is blocked. These observations together suggest a scenario whereby accumulation of the TBC1D15 oncoprotein drives deregulated TIC proliferation and formation of liver tumors due to alcohol-induced suppression of autophagy. This proposal resonates with accumulating evidence that suppression of autophagy, including through targeted genetic ablation of core autophagic machinery components, promotes the accumulation of oncoproteins leading to tumor formation [73–75], underscoring the importance of autophagic degradation in the tonic suppression of cancer. These findings suggest that depletion of p53 levels through aPKCζ activation and TBC1D15 upregulation may in turn cause de-repression of Tbc1d15 transcription, further accelerating p53 degradation and establishing a self-reinforcing feedback cycle. This cycle represents an attractive therapeutic target for inhibition of TICs in HCC, and developing a deeper understanding of the aPKCζ-NUMB-TBC1D15 regulatory axis in p53 degradation will further define optimal therapeutic targets.
More broadly, defining the machinery that controls the expansion of TICs will have important ramifications for cancer treatment. Conventional chemotherapy kills a large fraction of tumor cells, resulting in a transient reduction in tumor volume. However, it typically fails to eradicate TICs and may actually impose a strong selective pressure for TIC survival [76]. As a result, following chemotherapy tumors are often enriched with TICs resistant to subsequent treatments. To be effective in the long term, cancer therapies will need to include agents that target TIC survival and self-renewal. We propose that selective inhibition of the machinery that drives inappropriate, self-renewing, symmetrical divisions in TICs will lead to “sterilization” of the tumor and to a lasting, beneficial clinical response. The newly elucidated mechanistic framework for TIC proliferation described here represents an innovation that holds significant potential as a prospective therapeutic target.
8.7 Conclusions and Discussions
CD133+ TICs have previously been isolated from liver tumors of PTEN or MAT1A deficient mice [51, 52]. Using the same surface marker, we successfully isolated CD133+/CD49f+ TICs which activate a unique TLR4-NANOG pathway as an integral component for their self-renewal and tumorigenic activities. These TICs are also identified in HCC sections of alcoholic HCV patients by immunostaining [32] and isolated from such patients to validate induction of TLR4-dependent stemness genes and transformation [32]. These TICs respond to endotoxin to initiate tumorigenesis as shown in alcohol-fed HCV Tg mouse models, but TICs isolated from alcohol Core Tg mice and alcoholic HCV patients grow efficiently in vitro without addition of LPS but this growth is reduced by TLR4 knockdown, suggesting LPS-independent mechanisms of TLR4 activation in these cells which remain to be elucidated. Possibilities include non-LPS ligands such as HMGB1 released in inflammation activating TLR4 and protein–protein interactions leading to ligand-independent activation. Although we began and focused our studies on alcohol-HCV synergism, the oncogenic role of TLR4 has been extended to HCC of non-viral and non-alcohol etiology such as that in Spnb+/− mice and NAFLD patient [32]. A recent study demonstrates the critical role of endotoxin-activated TLR4 in promotion but not initiation of hepatocarcinogenesis induced by diethylnitrosamine and carbon tetrachloride [38]. We believe that the TLR4-dependent mechanisms of TIC generation actually contribute to or at least promote the initiation of HCC. A conceptual breakthrough of our findings is that the proinflammatory TLR4 is now considered as a putative proto-oncogene in hepatocarcinogenesis that links inflammation to carcinogenesis, the notion which has been entertained for the past 150 years. Based on this renewed concept, our studies have offered two novel insights into the molecular mechanisms of TLR4-mediated TICs’ tumorigenic activity (see a schematic diagram shown in Fig. 8.3): NANOG-dependent upregulation of IGF2BP3 and YAP1 which in turn block the TGF-β tumor suppressor pathway; and NANOG-mediated p53 degradation by disengagement from the protective NUMB protein via TBC1D15 interaction. These studies are now exploring potential mechanistic connections to metabolic programming known to occur in cancer cells and TICs in promoting and maintaining “stem cell fate” via molecular, genetic, and epigenetic mechanisms.
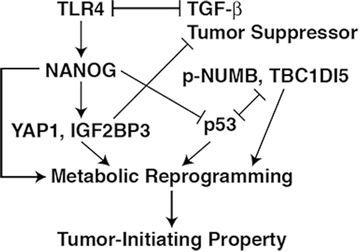
Fig. 8.3
 Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data
Acetaldehyde and Retinaldehyde-Metabolizing Enzymes in Colon and Pancreatic Cancers
Alcohol, Carcinoembryonic Antigen Processing and Colorectal Liver Metastases
Application of Mass Spectrometry-Based Metabolomics in Identification of Early Noninvasive Biomarkers of Alcohol-Induced Liver Disease Using Mouse Model
Alcohol Metabolism by Oral Streptococci and Interaction with Human Papillomavirus Leads to Malignant Transformation of Oral Keratinocytes
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data
Acetaldehyde and Retinaldehyde-Metabolizing Enzymes in Colon and Pancreatic Cancers
Alcohol, Carcinoembryonic Antigen Processing and Colorectal Liver Metastases
Application of Mass Spectrometry-Based Metabolomics in Identification of Early Noninvasive Biomarkers of Alcohol-Induced Liver Disease Using Mouse Model
Alcohol Metabolism by Oral Streptococci and Interaction with Human Papillomavirus Leads to Malignant Transformation of Oral Keratinocytes




TLR4-NANOG oncogenic pathway activated by alcohol, obesity, and HCV. TLR4 is ectopically activated in the liver by alcohol abuse, obesity, or/and HCV infection. This results in expression of the stem cell factor NANOG to mediate the genesis of TICs and tumorigenesis via antagonism of the TGF-β tumor suppressor pathway and degradation of p53 mediated by the oncoprotein TBC1D15. Upregulated NANOG and TBC1D15 and conversely suppressed p53 also contribute to anabolic metabolic reprogramming that is associated with the cancer stem cell fate regulation of TICs
Related posts:
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Alcohol and Breast Cancer: Reconciling Epidemiological and Molecular Data
Acetaldehyde and Retinaldehyde-Metabolizing Enzymes in Colon and Pancreatic Cancers
Alcohol, Carcinoembryonic Antigen Processing and Colorectal Liver Metastases
Application of Mass Spectrometry-Based Metabolomics in Identification of Early Noninvasive Biomarkers of Alcohol-Induced Liver Disease Using Mouse Model
Alcohol Metabolism by Oral Streptococci and Interaction with Human Papillomavirus Leads to Malignant Transformation of Oral Keratinocytes
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
