Tumor type
Prevalence (%)
Papillary carcinoma
BRAF
40–45
RET/PTC
10–20
RAS
10–20
TRK
<5
Follicular carcinoma
RAS
40–50
PAX8-PPARγ (PAX8-PPARG)
30–35
PIK3CA
<10
PTEN
<10
Poorly differentiated carcinoma
RAS
20–40
CTNNB1
10–20
TP53
20–30
BRAF
10–20
PIK3CA
5–10
PTEN
5–10
Anaplastic carcinoma
TP53
50–80
CTNNB1
5–60
RAS
20–40
BRAF
20–40
PIC3CA
10–20
PTEN
5–15
AKT1
5–10
Medullary carcinoma
Familial forms
RET
>95
Sporadic
RET
40–50
RAS
25
BRAF mutation is associated with aggressive tumor characteristics of PTC, such as extrathyroidal extension, lymph node and/or distant metastases, and advanced tumor stage at presentation [1, 22, 23] see Box 11.1. Additional evidence for more aggressive tumor behavior comes from the analysis of tumor stage. A significant proportion of BRAF-mutation positive tumors present at the advanced stage of the disease when compared with tumors that harbor the RET rearrangement, RAS mutation, and those with no mutations [1]. This can be attributed to the fact that BRAF mutation-positive patients are older and have a higher incidence of extrathyroidal extension and lymph node metastasis, the combination of which yields a higher tumor stage and consequently, less-favorable prognosis. Other studies have found an association between BRAF mutation and distant metastasis [10, 24]. More importantly, BRAF mutation has been found to be an independent predictor for resistance to radioactive iodine therapy and other treatment failures, and tumor recurrence even in patients with low-stage disease and as an independent risk factor for tumor-related death [4, 25–29].
The MAPK pathway plays a major role in tumor development and progression of PTC. Consequently, the MAPK pathway is a potentially effective therapeutic target for PTC. In recent years, a lot of work has been done to investigate the therapeutic potential of suppressing the pathway in human cancers. For thyroid cancer, recent effort has been particularly directed toward testing the therapeutic potential of knocking down the MAPK pathway signaling aberrantly activated by BRAF mutation. RAF kinase inhibitor AAL881 has been demonstrated to inhibit MEK and ERK phosphorylation and also induce apoptosis preferentially in cells that harbor BRAF V600E mutation when compared with wild-type ones, possibly because of better inhibitory activity against BRAF(V600E) [30]. Another inhibitor of RAF kinase activity, LBT-613 is also thought to hold promise for most forms of PTC. It blocks RAF-dependent MEK and ERK phosphorylation and also potently blocks MEK phosphorylation in human thyroid cancer cell lines with BRAF V600E mutations [31]. Efforts have also been made to develop strategies targeting MEK with anticancer drugs to suppress the MAPK pathway signaling. MEK inhibitor CI-1040 inhibits cell proliferation in thyroid cancer cells harboring BRAF V600E mutation [32].
The various small molecule inhibitors of activated BRAF serine/threonine kinase that have been developed are categorized on the basis of their mechanism of action. Type I inhibitors target a highly conserved motif containing a catalytic aspartate residue in the so-called “DFG-in” conformation, leading to interactions with the kinase hinge region and ATP binding site of the activated form of the kinase while Type II inhibitors bind to the ATP binding site itself and an adjacent hydrophobic pocket created by the activation loop. Type I inhibitors with preferential binding to the kinase domain of BRAF in the active conformation demonstrate greater inhibitory potency against the BRAF V600E mutant kinase than the wild-type [33]. In thyroid cancer cells where, RAF1 (also known as CRAF) expression is thought to be minimal, selective targeting of the mutant BRAF most associated with aggressive disease is a highly attractive therapeutic option [34]. Vemurafenib is a potent kinase inhibitor of BRAF V600E whose effect has been studied in patients with malignant melanoma. It inhibits proliferation and ERK phosphorylation in cell lines bearing activating mutations in codon 600 in a dose-dependent manner [35]. Preclinical studies in PTC have mimicked the experience with melanoma [36]. Regorafenib inhibits both wild-type and mutant BRAF and has been studied in a phase 1 clinical trial of continuous daily dosing that has been previously reported [37]. There was no objective response in any of the five thyroid cancer patients in the trial.
Fine needle aspiration (FNA) cytology is currently the most reliable diagnostic test in the evaluation of thyroid nodules [38, 39]. An estimated 20–30 % of FNA specimens present with equivocal cytological findings and are thus classified as indeterminate. Lesions in the indeterminate category include follicular lesions of undetermined significance/atypia of undetermined significance (FLUS/AUS), follicular neoplasm/suspicious for follicular neoplasm and suspicious for malignancy [40, 41]. Preoperative planning of optimal surgical management in patients with these cytologically indeterminate lesions may be challenging, with significant management implications ranging from delay in definitive surgery to unnecessary surgical intervention. To avoid this dilemma, a number of studies have evaluated the use of cytological preparations for BRAF V600E molecular analysis and they have shown that it improves the accuracy of cytologic diagnosis of thyroid nodules [42–48]. In view of the high specificity of BRAF mutation in the detection of PTC, indeterminate lesions with BRAF mutation are triaged for total thyroidectomy with or without central lymph node dissection. This eliminates the need for second surgery in up to 40 % of patients with indeterminate cytologic diagnosis. For patients with follicular neoplasm and suspicious for malignancy diagnoses and are BRAF mutation negative, lobectomy with or without intraoperative consultation is indicated. For BRAF mutation negative FLUS/AUS lesions, patients may be followed up conservatively for 6–12 months unless there are other concomitant risk factors [42, 43, 49]. It has been shown that the presence of preoperative detection of BRAF V600E mutation is perhaps the only available preoperative clinical parameter, which independently predicts central compartment lymph node metastasis in patients with PTC. This further supports BRAF mutation testing in the preoperative setting thereby guiding the extent of initial surgical management for patients with PTC [50–52].
Exon 15 nucleotide alterations account for approximately 90 % of all BRAF mutations in thyroid cancer and the additional 10 % of mutations involve exon 11. Ideally, a comprehensive diagnostic assay should be optimized to detect mutations within both these regions. However, for diagnostic purposes, more than 95 % of clinically relevant mutations can be covered by assays examining exon 15, particularly the V600E mutation. In clinical practice, almost all available methodologies are PCR-based assays and essentially any type of tissue or cytological material can be used for BRAF mutation testing. BRAF mutation testing requires a much higher analytical sensitivity and specificity in dealing with cytological specimens where the tumor cells may be a minor population in a background of non-neoplastic cells. While PCR-Sanger sequencing, allelic-specific amplification, melting curve analysis [53, 54], real-time PCR are common laboratory methods, BRAF mutation detection methods that have the highly desired sensitivity and specificity appropriate for testing minimal tissue or cytological samples include shifted termination primer extension assay (STA), single-strand conformation polymorphism (SSCP) analysis, pyrosequencing, and coamplification at low denaturation temperature—PCR (COLD-PCR) [55].
Single-strand conformation polymorphism (SSCP) analysis detects randomly distributed mutations. It is the traditional method for high-sensitivity detection of a known point mutation [56–58]. When combined with follow-up Sanger sequencing of abnormal SSCP products, the method is able to direct variant mutations within the amplicon with high sensitivity and specificity. DNA is extracted and amplified using primers flanking BRAF exon 15. PCR products are then analyzed by polyacrylamide or capillary gel electrophoresis. The SSCP band pattern is compared to that of a positive BRAF V600E DNA control sample, for interpretation. If abnormal bands are present but do not match those of the positive control, the abnormal SSCP bands are purified from the gel and Sanger sequencing is then performed to identify the variant mutations of exon 15 [55]. The drawback of this method is that it does not allow identification of the precise nucleotide change and requires an additional complementary method. In addition, the method is also labor intensive.
More recently, the use of VE1 immunohistochemistry in the assessment of BRAF mutational status in thyroid core needle biopsy has been investigated [59]. The results demonstrated good concordance between immunophenotypical VE1 expression and V600E mutational status. Homogeneous BRAF immunohistochemical staining occurs in the vast majority of cases and absent/faint staining for BRAF correlates with the lack of BRAF V600E mutation. However, only strong immunohistochemical staining is highly correlated with the BRAF mutation in thyroid carcinomas including papillary, poorly differentiated, and anaplastic histological types. Moderate staining cannot be relied on and molecular testing should be performed to confirm the presence of BRAF mutations [60].
RET/PTC Rearrangement
RET (also known as RET/PTC) rearrangement is another diagnostic marker of PTC. However, the detection of this alteration is more difficult and the interpretation can be challenging because the distribution of the rearrangement within the tumor can be heterogeneous and vary from being clonal (involving most neoplastic cells) to being nonclonal (detected in a very small fraction of tumor cells) [61–63]. The heterogeneity may be of a potential problem for the RET receptor-targeted therapy, since tumors with nonclonal RET frequently have other genetic alterations and may not respond to RET inhibitors in the same way as tumors harboring the clonal rearrangement.
The RET proto-oncogene is located on chromosome 10q11.2 and encodes for a transmembrane tyrosine kinase receptor involved in the control of cell differentiation and proliferation [64]. It is highly expressed in thyroid parafollicular cells but not in follicular cells. In follicular cells, RET can be activated by chromosomal rearrangement, which enables the RET protein to activate the MAPK signaling pathway [5, 65–69]. As a result of the rearrangement, a portion of the RET gene is fused to one of several possible partner genes. It is formed by the fusion between the 3′ portion of the RET receptor tyrosine kinase gene and the 5′ portion of various unrelated genes [1]. All chimeric genes contain the portion of RET that encodes the intact tyrosine kinase domain fused to an active promoter of another gene that drives the expression and ligand-independent dimerization of the RET protein, which leads to chronic stimulation of MAPK signaling and tumorigenesis in thyroid cells [2, 67–69]. More than ten types of RET rearrangement have been reported [4, 70, 71] (Table 11.2). The two most common types of rearrangement, RET with CCDC6 (RET/PTC1)) and RET with NCOA4 (RET/PTC3) [66, 72] are paracentric, intrachromosomal inversions as all fusion partners reside on the same long arm (“q”) of chromosome 10 [73, 74]. By contrast, the other types of RET (RET/PTC) rearrangements are all interchromosomal rearrangements formed by RET fusion to genes located on different chromosomes [75–83].
Table 11.2
Different types of RET (RET/PTC) rearrangements found in papillary thyroid carcinomas
RET (RET/PTC) type | Partner gene | Chromosomal positions |
|---|---|---|
RET/PTC1 | CCDC6 (H4) | 10q21 |
RET/PTC2 | PRKAR1A | 17q23 |
RET/PTC3 | NCOA4 (ELE1) | 10q11.2 |
RET/PTC4 | NCOA4 (ELE1) | 10q11.2 |
RET/PTC5 | GOLGA5 | 14q |
RET/PTC6 | TRIM24 | 7q32–34 |
RET/PTC7 | TRIM33 | 1p13 |
RET/PTC8 | KTN1 | 14q22.1 |
RET/PTC9 | RFG9 | 18q21–22 |
ELKS/RET | ELKS | 12p13.3 |
PCMI/RET | PCM1 | 8p21–22 |
RFP/RET | TRIM27 | 6p21 |
HOOK3/RET | HOOK3 | 8p11.21 |
Box 11.2: Characteristics of Papillary Carcinoma with RET/PTC Rearrangement
Classic papillary and diffuse sclerosing variant
Young age at presentation
Association with radiation exposure
High rate of lymph node metastasis
Lower stage at presentation
RET (RET/PTC) rearrangement is found on average in 10–20 % of adult sporadic papillary carcinomas [62, 84]. RET rearrangement has significant geographic variation and it occurs with higher incidence in patients with a history of radiation exposure (50–80 %) and in papillary carcinomas from children and young adults (40–70 %) [85–88]. RET-CCDC6is typically the most common rearrangement type and make up about 60–70 % of all positive cases. RET-NCOA4 (RET/PTC3) accounts for 20–30 % while RET-PRKAR1A (RET/PTC2) and other arrangement types account for less than 5 % of all detected rearrangements [89, 90].
Papillary carcinomas with RET rearrangement typically present at younger age and have classic papillary architecture and a high rate of lymph node metastases [1] see Box 11.2. The rearrangement particularly RET-CCDC6 type, is typically found in papillary carcinomas, with classic papillary architecture, but can also be seen in papillary microcarcinomas and other microscopic variants of papillary carcinoma, most notably the diffuse sclerosing variant [90–92]. In tumors arising after radiation exposure, RET-CCDC6 is associated with classic papillary histology, and RET-NCOA4 (RET/PTC3) type, with the solid variant of papillary carcinoma [85].
Correlation between RET rearrangement and prognosis in PTC is unclear. Some existing evidence suggests that RET-CCDC6 rearrangement is associated with more favorable behavior of PTC. When compared to tumors harboring BRAF and RAS mutations, tumors harboring RET-CCDC6 have a very low probability of progression to poorly differentiated and anaplastic carcinoma [88, 90, 93]. It has also been suggested that tumors carrying RET-NCOA4 (RET/PTC3) rearrangement may be prone to dedifferentiation and more aggressive behavior [92, 94].
It has been demonstrated that MEK1/2 inhibitors can inhibit PTC cell growth in-vitro and the expression of phosphorylated ERK1/2 decreased in PTC cells with the RET-CCDC6 rearrangement [95, 96]. Since the approval of Sorafenib, a multikinase inhibitor by the U.S. Food and Drug Administration for treating advanced renal cell carcinoma, there has been many Phase I, II, and III trials for different types of cancer [95, 97]. Sorafenib has been found to be more potent in PTC cells with RET-CCDC6 rearrangement than in those with a BRAF mutation, hence it may be an ideal drug for selected PTC patients with RET rearrangement. SU11248 (Sunitinib), a selective, orally administered receptor tyrosine kinase (RTK) inhibitor that targets platelet-derived growth factor receptor (PDGFR), vascular endothelial growth factor receptor (VEGFR), and fms-related tyrosine kinase 3 (FLT3) has been shown to be a potent inhibitor of RET oncoproteins. SU-11248-mediated block of RET inhibit its signaling, decrease RET autophosphorylation, and STAT3 activation and block the transforming capacity of RET [98, 99].
Clonal RET (RET/PTC) is highly specific for PTC. It has been suggested that detection by RT-PCR or FISH in surgical or FNA samples has a positive predictive value of 100 % in PTC. The choice of the detection method is dictated by the type of sample available. Reliable diagnostic methods for the clinical detection include (1) reverse transcriptase PCR (RT-PCR) using RNA isolated from snap frozen tumor tissue or FNA sample in a conventional or real-time mode with a single round of amplification and with cut-off levels for detection established to ensure that at least 1 % of tumor cells carry the rearrangement [100], and (2) fluorescence in-situ hybridization (FISH) with appropriate cut-off levels that can be performed on formalin-fixed and paraffin-embedded tissues [61]. This assay should be validated to set up appropriate cut-off levels, which generally should be no fewer than 8–12 % of cells with the rearrangement pattern of signals to avoid detection of nonclonal rearrangements [4, 62].
While studies that have used Southern blot analysis and regular sensitivity RT-PCR found no RET rearrangement in benign thyroid tumors [90, 101–103], other analyses based on highly sensitive methods have identified the presence of RET rearrangements in 10–45 % of thyroid adenomas and in other benign nodules and non-neoplastic thyroid lesions [104–107]. These RET rearrangements are nonclonal, as they occur in a small proportion of cells within the tumor nodule or even in single non-neoplastic thyroid cells. Some studies have reported the occurrence of RET rearrangement in hyalinizing trabecular tumors. These findings provide evidence suggesting that hyalinizing trabecular tumors represent a peculiar variant of PTC. However, these studies did not investigate whether or not RET was present in the majority of cells within these tumors and therefore cannot provide conclusive biological evidence for linkage between papillary carcinoma and hyalinizing trabecular tumor [3, 108–110].
The diagnostic impact of RET (RET/PTC) in surgical material is limited because most of RET-positive tumors are either classic papillary carcinoma or diffuse sclerosing variants. However, it may have a high diagnostic value in thyroid FNAs. Several studies have shown that RET detection can refine the preoperative diagnosis of thyroid nodules, particularly in samples that are indeterminate by cytology or have insufficient amount of cells for routine cytologic evaluation [48, 111, 112].
Using Immunohistochemistry (IHC) assay, a correlative trend has been observed between the degree of RET IHC staining and RET rearrangement in that the majority of moderately stained cases were RET rearranged while most of the weakly stained cases were RET non-rearranged [113]. Although RET IHC is not routinely used, this suggests that with further validation and optimization, IHC could offer an alternative detection methodology for RET rearrangements.
RAS Mutation
RAS represents a family of small guanosine triphosphate (GTP)-binding proteins (G proteins) that are essential signaling molecules downstream of many cell surface receptors. Activation of RAS results in a cascade of signaling activity, primarily through the RAF-MEK-ERK cascade that regulates a wide variety of cellular functions, including cell cycle progression, proliferation, motility, and apoptosis [114]. HRAS, KRAS, and NRAS genes encode highly related G-proteins that are located at the inner surface of the cell membrane and transmit signals arising from cell membrane receptor tyrosine kinases and G-protein-coupled receptors along the MAPK, PI3K-AKT, and other signaling pathways. In its inactive state, RAS protein is bound to guanosine diphosphate (GDP). Upon activation, it releases GDP and binds guanosine triphosphate (GTP), activating the MAPK and other signaling pathways, such as PI3K-AKT [2, 115]. Activating point mutations typically affect codons 12, 13, and 61 of the RAS genes. In thyroid cancer, NRAS codon 61 and HRAS codon 61 mutations are the most common. Testing for these two hot spots detects the vast majority of RAS mutations [2].
Box 11.3: Characteristics of Papillary Carcinoma with RAS Mutation
Follicular variant of papillary carcinoma
Less-prominent nuclear features
More frequent distant metastasis
Point mutations of the RAS gene are not restricted to any particular type of thyroid tumor. They are found with variable frequency in all types of thyroid follicular cell-derived tumors. In papillary carcinomas, RAS mutations occur in 15–20 % of tumors [116–121]. Papillary carcinomas harboring RAS mutation are almost always the follicular variant of PTC (FVPTC) but this mutation is rare in classic papillary carcinoma (Table 11.3). RAS mutation also correlates with significantly less-prominent nuclear features of papillary carcinoma, more frequent encapsulation, and low rate of lymph node metastases [1, 3, 122] see Box 11.3. RAS mutations are also found in 40–50 % of conventional type follicular carcinomas [119, 123–127], and 20–40 % of conventional type follicular adenomas [116, 123–126]. In adenomas, the mutations appear to be more common in tumors with a microfollicular growth pattern [123]. A lower incidence has been reported in oncocytic tumors, in which only 0–4 % of adenomas and 15–25 % of carcinomas appear to be affected [125, 128, 129] (Table 11.4). Some studies have reported RAS mutations in hyperplastic nodules; however, since these lesions carry a clonal mutation, it is likely that these lesions are true neoplasms and therefore should be designated as follicular adenomas, despite their frequent macrofollicular colloid-rich histology [3].
Table 11.3
Prevalence of mutations in classic and follicular variant of papillary thyroid carcinoma
BRAF (%) | RET/PTC (%) | RAS (%) | |
|---|---|---|---|
Classic papillary carcinoma | 55 | 25 | 0 |
Follicular variant | 5 | <5 | 45 |
Table 11.4
Prevalence of RAS mutation in different tumor types
Tumor type | Prevalence (%) |
|---|---|
Follicular carcinoma | 40–50 |
Follicular adenoma | 20–40 |
Oncocytic carcinoma | 15–25 |
Oncocytic adenoma | 0–4 |
The presence of RAS mutation in follicular carcinomas has been found to correlate with tumor dedifferentiation and a less-favorable prognosis [18, 119, 130]. Several studies have found a significant correlation between RAS mutation and metastatic behavior of follicular carcinomas, especially with respect to bone metastases [117, 119, 131]. The more aggressive biological properties of these tumors may be due to the effect of the mutant RAS protein on promoting chromosomal instability, which has been demonstrated at least in the in vitro setting [132, 133]. The increasing instability may predispose the tumor cells to acquiring additional mutations, which would result in more malignant phenotype.
The high-grade thyroid neoplasms with RAS mutations are encountered much less frequently and are represented by neoplasms such as poorly differentiated carcinoma, anaplastic carcinoma, and medullary carcinoma. These neoplasms are genotypically complex and poorly differentiated carcinoma and anaplastic carcinoma often demonstrate additional mutations in TP53 (p53); CTNNB1; phosphoinositide-3-kinase, catalytic, alpha (PIK3CA); phosphatase and tensin homolog (PTEN); and/or AKT1 genes. In some cases, foci of poorly differentiated carcinoma or anaplastic carcinoma may have arisen from well-differentiated cancers such as FVPTC. Therefore, the RAS mutation appears to represent an early event in thyroid tumor progression [134].
Farnesylation of RAS proteins is necessary for their activity and is catalyzed by a family of enzymes called farnesyl protein transferases (FPTases). Thus, FPTases represent a potential therapeutic target for inhibiting Ras-mediated signaling in cancer cells [114]. Manumycin, a natural product of streptomyces, has been shown to possess antineoplastic properties through inhibition of FTPase activity. In vitro, manumycin reduces cell growth and survival of six anaplastic thyroid cancer cell lines as a solitary agent and in a cooperative manner with paclitaxel, doxorubicin, or cisplatin, agents with a modest antitumor effect in anaplastic thyroid carcinoma [135]. The combination of manumycin plus paclitaxel was further tested in vivo, where it inhibited growth and angiogenesis in a mouse xenograft model of anaplastic thyroid carcinoma [136]. Manumycin has not been evaluated in clinical trials because of toxicity in animal models; however, other FTPase inhibitors, such as R115777 (Tipifarnib) and SCH66336 (Lonafarnib), are being studied alone and in combination for other malignancies.
The CI-1040 compound is a potent dual inhibitor of both MAP2K1 (MEK1) and MAP2K2 (MEK2) that exhibits significant antitumor activities in in vitro and in vivo studies [137, 138]. The therapeutic potential of CI-1040 for thyroid cancer has been studied. Its effects were examined on a large number of human thyroid tumor cell lines with different genotypes in the MAPK pathway and it was demonstrated that CI-1040 selectively inhibited proliferation of thyroid cancer cells and their xenograft tumors with BRAF and RAS mutations but not cells without these genetic alterations. Therefore, BRAF and RAS mutations, through activating the MAPK pathway, both confer thyroid cancer cells sensitivity to the MEK inhibitor. Interestingly, CI-1040 virtually had no effect on the proliferation of PTC-derived cells that harbored RET-CCDC6 (RET/PTC1) rearrangement [32].
The diagnostic use of RAS mutation detection is controversial. On the one hand, it is not specific for malignancy since RAS mutations also occur with significant prevalence in benign follicular adenomas. On the other hand, RAS mutations frequently occur in follicular carcinomas and the follicular variant of papillary carcinomas, both of which are difficult to diagnose cytologically in thyroid FNA samples. Moreover, since mutant RAS is likely to predispose to progression from follicular adenoma to carcinoma and to further tumor dedifferentiation, it may be justifiable to surgically remove the RAS-positive adenomas to prevent such a progression. In a prospective study aimed at assessing the role of detection of different mutations in improving the preoperative FNA diagnosis of thyroid nodules, the detection of RAS mutations was found to improve the diagnostic accuracy and allowed to diagnose malignant tumors in several samples with negative or insufficient cytology [139].
Point mutations in codons 12/13 and 61 of the HRAS, KRAS and NRAS genes can be detected using different methods. LightCycler PCR and fluorescence melting curve analysis (FMCA) is very commonly used. LightCycler technology has been demonstrated as an efficient method for detection of NRAS mutations in human tumors [140–142]. It is based on rapid-cycle PCR amplification of the locus containing a mutational hot spot on the LightCycler using a hybridization probe format, followed by FMCA. This format requires a pair of primers and two fluorescently labeled oligonucleotide probes and relies on fluorescence resonance transfer interaction between two probes that hybridize adjacent to one another, so that one fluorophore functions as the donor and another as the acceptor, the latter emitting a light of specific wavelength [140, 143]. For the detection of a point mutation, one probe is designed to span the mutation site, and the products are distinguished based on their distinct melting temperatures, which reflect the thermodynamic stability of the perfectly complementary and mismatched probe-target duplexes. For each mutational hot spot, a pair of primers and two internal oligonucleotide probes are used. Amplification is performed followed by post-amplification. Fluorescence melting peaks are built by plotting of the negative derivative of fluorescent signal corresponding to the temperature. All PCR products that show deviation from the wild-type melting peak are sequenced after purification to verify the presence of RAS mutation and detect the exact nucleotide change.
Another method is by DNA heteroduplex analysis, a method for identification of randomly distributed mutations. After amplification of the DNA sequence of interest, double-stranded PCR products are denatured by heat and slowly cooled to achieve random reannealing of the single-stranded DNA molecules and loaded into a gel for electrophoresis. The presence of mismatched based pairs between the strands in heteroduplexes (strands with a mutation) changes their structural conformation and forces them to migrate more slowly in a gel than homoduplexes (strands with no mutation), providing evidence for a mutation [100]. Direct DNA sequencing can also be used for the detection of heterozygous mutations in the RAS genes.
PAX8-PPARγ (PAX8-PPARG) Rearrangement
Paired box gene 8-peroxisome proliferator-activated receptor gamma (PAX8-PPARG) rearrangement is a molecular event associated with follicular thyroid tumorigenesis and is generated by a chromosomal rearrangement between PAX8 and PPARγ (PPARG) genes. It is a somatic genetic rearrangement, involving a 3p25 and 2q13 translocation [144, 145]. This translocation t(2;3)(q13;p25) creates a fusion gene, between the transcription factor paired box gene 8 (PAX8) and the nuclear receptor peroxisome proliferator-activated receptor gamma (PPARG). The PAX8-PPARG fusion protein is expressed in thyroid cells under the control of the PAX8 promoter, which is active in thyroid follicular cells [146]. PAX8 has a key role in thyroid cell differentiation and growth [147], whereas PPARG is a ligand-dependent transcription factor that is activated by the thiazolidinedione class of antidiabetic drugs [148–150], and has a fundamental role in adipogenesis and glucose metabolism.
Box 11.4: Characteristics of Tumors with PAX8-PPARG Rearrangement
Younger age at presentation
Smaller size
Solid/nested growth pattern
Frequent vascular invasion
The PAX8-PPARG fusion gene has been identified in up to 35 % of conventional follicular carcinomas and has a lower prevalence in Hürthle cell carcinomas [18, 151]. PAX8-PPARG has also been detected also in some follicular thyroid adenomas, albeit less frequently [140, 151–153]. It has been suggested that follicular adenomas positive for this rearrangement may in fact be preinvasive follicular carcinomas or tumors where invasion was overlooked during histological examination [140]. Other studies have reported the detection of PAX8-PPARG in some follicular variants of PTC [154, 155]. The oncogenic action of PAX8-PPARG could be related to the constitutive overexpression of full-length PPARG domain, interference with wild-type PPARG function, alterations of PAX8 function, formation of novel fusion gene activities, or combination of such events [145].
Tumors harboring PAX8-PPARG tend to present at a younger age, be smaller in size, have a solid/nested growth pattern and more frequently show vascular invasion when compared with follicular carcinomas that are negative for this rearrangement [140, 154]. See Box 11.4.
The mechanisms of cell transformation induced by PAX8-PPARG are not fully understood and this lack of understanding hampers its use as a therapeutic target in follicular carcinomas [18]. PPARG is one of three isoforms of PPAR, and regulates cellular growth and differentiation. The effects of PPARG agonists on cancer cells include growth inhibition, induction of apoptosis, and redifferentiation. Recent studies have examined the effects of PPARG agonist treatment in a series of thyroid cancer cell lines [114]. Ohta and colleagues [156] found that in four PPARG-positive papillary thyroid cancer cell lines, treatment with troglitazone (10 μmol/L) and rosiglitazone (10 μmol/L) inhibited growth and stimulated apoptosis by means of the c-myc pathway; there was no effect on growth or apoptosis in the PPARG-negative thyroid cancer cell lines. In vivo studies with tumor xenografts in nude mice resulted in significant growth inhibition when troglitazone treatment was initiated. Martelli and colleagues [157] showed that transfection of wild-type PPARG cDNA into the PPARG-negative papillary thyroid cancer cell line resulted in a significant growth inhibitory effect when ciglitazone treatment was initiated. The growth inhibitory effects of ciglitazone in these experiments seemed to be related to cell cycle arrest mediated by p27. Flow cytometry analysis of ciglitazone-treated cells showed a significant increase in the percentage of cells undergoing apoptosis. Currently, there is a clinical trial of rosiglitazone in advanced or metastatic thyroid cancer.
The detection of PAX8-PPARG rearrangement can be achieved by reverse transcriptase-PCR (RT-PCR) or FISH. Snap-frozen tissue or formalin-fixed paraffin-embedded blocks can be used for RT-PCR. RNA extraction is performed and all cDNA samples are tested to assess the adequacy of extracted RNA by amplifying a 247-bp control sequence of the PRKG1 (PGK) gene. To detect PAX8-PPARG fusion, PCR primers are designed taking into account that several RNA transcripts are often present within follicular carcinomas, including those corresponding to the fusion of exons 1–7, or 1–8, or 1–9, or 1–7 plus 9 of PAX8 to PPARG exons 1–6. Because RNA extracted from paraffin-embedded tissue is considerably degraded and can be used to amplify only short sequences, three upstream PAX8 primers located in exons 7, 8, and 9 are used paired with a downstream primer in exon 1 of PPARG. This accounts for all possible splicing variants so that the smallest target sequence amplified would be 67–69 bp long. PCR products are resolved by electrophoresis in a 1.5 % agarose gel. The amplified bands are cut out from the agarose gel, purified and sequenced [153]. PAX8-PPARG rearrangement results in overexpression of the PPARG protein so it can also be detected by immunohistochemistry. However, only strong diffuse nuclear staining correlates with the presence of rearrangement [18, 153]. Detection of PAX8-PPARG rearrangement in a follicular lesion is not fully diagnostic for malignancy by itself, but it should prompt an exhaustive search for vascular or capsular invasion [4]. PAX8-PPARG rearrangement can be detected in thyroid FNA samples, and this typically correlates with the presence of malignancy [47, 158].
RET Point Mutations
The RET receptor activates signaling pathways responsible for cell proliferation, survival, differentiation, motility, and chemotaxis [159]. Alteration of the RET proto-oncogene plays a causal role in the familial forms of medullary thyroid carcinoma and has also been found in the sporadic form of the disease [115]. Germline mutations in the discrete functional regions of RET are found in almost all patients with familial forms of medullary carcinoma. The RET protooncogene is located in the pericentromeric region of chromosome 10q11.2 and is composed of 21 exons [115, 160]. RET encodes a receptor tyrosine kinase, which is expressed in neuroendocrine cells and neural cells. Point mutations in RET protooncogene cause multiple endocrine neoplasia (MEN) 2A, MEN2B, and familial medullary thyroid carcinoma (FMTC). MEN2A is associated with mutations involving the extracellular cysteine codons 609, 611, 618, 620 (exon 10), 630, or 634 (exon 11). The mutations associated with FMTC involve a broad range of codons including some associated with MEN2A, particularly 609, 618, and 620, as well as others such as 768, 790, and 791 (exon 13), 804, 844 (exon 14), or 891 (exon 15). In 95 % of patients with MEN2B there is a point mutation in codon 918 (exon 16, Met918Thr) within the intracellular domain of RET [161].
In patients with MEN2A, MEN2B, and FMTC there is a correlation between genotype and phenotype, both in the clinical expression of the disease spectrum and the clinical aggressiveness of MTC. Whereas, virtually all patients develop MTC, pheochromocytomas develop in approximately 50 % of patients with MEN2A and MEN2B. In MEN2A they occur most often in patients with RET mutations in codon 634 and less frequently in patients with mutations in codons 618, 620, or 791. Hyperparathyroidism occurs only in patients with MEN2A who have mutations in codons 618, 630, 634, and 791. Considering the age of onset and clinical aggressiveness of MTC, patients are classified as being at highest risk, higher risk, or high risk. Patients with MEN2B, who have mutations in codons 918 or 883 represent the highest risk group, whereas patients with mutations in codons 634, 630, 609, 611, 618, and 620 are in the higher risk group. The high risk group includes patients with mutations in codons 768, 790, 791, 804, and 891 [161, 162].
ZD6474, an orally active low-molecular-weight receptor tyrosine kinase inhibitor, is a potent inhibitor of the kinase insert domain receptor (KDR or VEGFR2) and has been shown to be a potent inhibitor of RET oncoproteins [161]. Phase I studies showed that ZD6474 administration is associated with minimal toxicity and subsequent phase II clinical trials showed that ZD6474 has activity in patients with locally advanced or metastatic hereditary MTC. Approximately 20 % of patients experienced a partial remission, as shown by RECIST (response evaluation criteria in solid tumors) criteria and another 60 % experienced stable disease for a disease control rate of 80 % [163]. Following treatment, there was reduction in serum levels of calcitonin and carcinoembryonic antigen, tumor markers secreted by the MTC cells. Subsequently it has been shown to induce confirmed partial remissions in 85 % of children with MEN2B [164]. Phase II clinical trials of other molecular targeted therapeutic agents have also shown activity in patients with advanced MTC [165, 166].
For detection of point mutations in the RET gene, the most commonly used technique is direct nucleotide sequencing [167, 168]. All exons known to harbor mutations are amplified in several PCR reactions, directly sequenced and analyzed for mutation. If the location of the RET mutation is known from a test performed on other family members, the presence of this mutation can be tested for by PCR-RFLP [169]. Other techniques that can be used include SSCP and heteroduplex analysis, although they tend to have lower sensitivity and are more labor intensive [170, 171].
Other Genetic Alterations
NTRK1 (TRK) Rearrangement
The neurotrophic tyrosine kinase, receptor, type 1 (NTRK1 also known as TRK) proto-oncogene is located on chromosome 1q22. It encodes a tyrosine kinase receptor for nerve growth factor. In an exactly analogous fashion to RET, it undergoes constitutive activation by gene rearrangement. At least three types of the rearrangement exist, formed by the fusion of the NTRK1 gene to different partners. Two of them, the TPM3 gene and the TPR gene, are also located on chromosome 1q, and therefore these fusions are intrachromosomal inversions. The third fusion partner, the TFG gene, resides on chromosome 3, and this fusion is a result of the t(1,3) translocation. All fusion types lead to expression and activation of the tyrosine kinase domain of NTRK1 [89, 172–174]. The fusion is tumorogenic for thyroid follicular cells, as TPR-NTRK1 drives the development of papillary carcinoma. NTRK1 (TRK) rearrangement is a rare type of somatic mutation found in papillary carcinoma. It is found in less than 5 % of papillary carcinomas in most studies [175, 176], and with higher prevalence in some regions of the world [89, 172, 177]. The diagnostic utility of testing for NTRK1 (TRK) rearrangement is limited due to low prevalence of the alteration in thyroid tumors [178, 179].
TP53 and CTNNB1 Mutations
TP53 and CTNNB1 (β-catenin) are additional genes mutated in thyroid cancer. TP53 is a tumor suppressor gene that plays important roles in cell cycle regulation and DNA repair. Inactivating point mutations of the TP53 tumor suppressor gene are among the most common mutations found in human cancer. In thyroid tumors, TP53 mutations are a late event, reported in 60–80 % of anaplastic carcinomas and 15–30 % of poorly differentiated carcinomas [18, 180–182]. TP53 mutations have also been found in up to 22 % of oncocytic follicular carcinomas. Most of them involve exons 5–8 of the gene and alter its DNA-binding properties. CTNNB1 is involved in wingless (Wnt) signaling pathway. This gene tends to be mutated in a significant proportion of poorly differentiated and anaplastic carcinomas [183, 184]. These mutations in exon 3 disrupt the formation of a complex with GSK3β, apc, and axin, which leads to phosphorylation, ubiquitination, and degradation of β-catenin. These mutations lead to increased β-catenin signaling and nuclear localization and are not found in well-differentiated thyroid cancers [185].
PI3K/AKT Pathway Mutations
PI3K/AKT pathway is a key regulator of cell proliferation and inhibitor of apoptosis. Point mutations affecting PIK3CA and PTEN are found with some frequency in anaplastic carcinomas [18, 186–188]. Inactivating mutations in PTEN are seen in Cowden’s disease, a familial tumor syndrome associated with follicular carcinoma.
NDUFA13 Mutations
Mutations of the NDUFA13 gene (also known as GRIM19) have been identified in oncocytic thyroid tumors [2, 189]. This gene encodes a protein that regulates cell death and promotes apoptosis, and also affects mitochondrial metabolism by serving as an essential component of complex I of the mitochondrial respiratory chain [190]. The role of NDUFA13 mutations in carcinogenesis remains obscure [2].
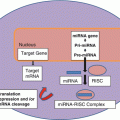
Emerging Tests in Diagnosis and Management
Other approaches are beginning to emerge in the management of thyroid lesions in the indeterminate categories and these may be the direction of management of thyroid nodules for the foreseeable future. There are tests utilizing gene expression or miRNA profiling to triage indeterminate thyroid FNAs [191]. These tests have a high negative predictive value, similar to the probability of malignancy for an initial benign diagnosis. Genetic tests based on a comprehensive panel of point mutations and gene fusions occurring in thyroid cancers seem to be the future of thyroid molecular diagnostics. This analysis is performed on a proprietary sequencer using the targeted ThyroSeq next generation sequencing panel, which simultaneously tests for point mutations in 13 genes and for 42 types of gene fusions that occur in thyroid cancer [192]. This has led to increasing the sensitivity of cancer detection to 90 % and a negative predictive value of 96 % (Table 11.5).
Table 11.5
The most commonly encountered genetic alterations in the thyroid
Assay/genetic abnormalities
Related posts:Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|
|---|