Thrombotic microangiopathies (TMA) such as atypical hemolytic uremic syndrome (aHUS) have evolved from rare, fulminant childhood afflictions to uncommon diseases with acute and chronic phases involving both children and adults. Breakthroughs in complement and coagulation regulation have allowed redefinition of specific entities despite substantial phenotypic mimicry. Reconciliation of phenotypes and delivery of life saving therapies require a multidisciplinary team of experts. The purpose of this review is to describe advances in the molecular pathophysiology of aHUS and to share the 2014 experience of the multidisciplinary Johns Hopkins TMA Registry in applying diagnostic assays, reporting disease associations, and genetic testing.
Key points
- •
Thrombotic microangiopathies (TMA) are a diverse group of diseases with distinct as well as overlapping pathophysiologic mechanisms.
- •
TMA is a multifactorial disease, and depends on the relative strengths of constitutional predisposition and external triggers.
- •
Atypical hemolytic uremic syndrome is characterized by dysregulated activity of the alternative pathway of complement for which blockade at C5 is an effective therapy.
- •
Owing to heterogeneity in pathophysiology, therapy for TMA must be individualized. An individual patient may exhibit multiple mechanisms of disease.
Case presentation
A 31-year-old female presented at 20.2 weeks gestation with fever, dyspnea, acute kidney injury (creatinine, 1.9 mg/dL), dusky painful fingertips, red cell fragmentation, and a platelet count of 7000 cells/mm 3 . On the second hospital day, she delivered a nonviable fetus. Continuous venovenous hemodialysis was initiated, and laboratory studies were notable for mildly prolonged coagulation times, hypofibrinogenemia, and d -dimer greater than 30 mg/L. A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity was 41% (normal, 70%–155%) and complement C3 and C4 were normal. Despite improving with supportive care, she developed seizures on hospital day 10. The C3 was now low at 70 mg/dL (normal, 79–152), and therapeutic plasma exchange (TPE) was initiated. Over the next 2 weeks, she received daily TPE with recovery of both platelets and renal function, and ADAMTS13 activity normalized. After discontinuation of TPE, however, platelets and renal function worsened, and ADAMTS13 activity fell to 19%. TPE was reinitiated, and rituximab was administered. Renal biopsy obtained 48 days after her presentation revealed focal cortical infarction, congested glomeruli with loss of capillary endothelial cells, mesangiolysis with embedded erythrocyte fragments, and intimal edema with endothelial activation in an interlobular artery. Immunofluorescence was notable for segmental granular mesangial immunoglobulin (Ig)A (sparse 1–2+), IgM (1+), C3 (1+), kappa (1–2+), and lambda (sparse trace – 1+). Segmental capillary wall staining for C3 (1–3+) was present, and electron microscopy revealed subendothelial widening, capillary loop collapse, and endothelial cell swelling with no electron dense deposits. Platelets and renal function normalized after 29 plasma exchanges. Relevant serologic testing for antinuclear antibodies, antiphospholipid antibody syndrome, and human immunodeficiency virus was negative. Nine months after her presentation, genetic testing identified a heterozygous pathogenic mutation in thrombomodulin ( THBD ; c.1456G>T; p.Asp486Tyr), and a previously unreported heterozygous mutation in complement factor H related protein-5 ( CFHR5 ; c.1357C>G; p.Pro453Ala) of unknown significance, consistent with a diagnosis of atypical hemolytic uremic syndrome. Two years later she remains in remission with serum creatinine 0.7 mg/dL (estimated glomerular filtration rate, >60 mL/min/1.73 m 2 ), normal urine protein excretion, and normal hematologic parameters.
Case presentation
A 31-year-old female presented at 20.2 weeks gestation with fever, dyspnea, acute kidney injury (creatinine, 1.9 mg/dL), dusky painful fingertips, red cell fragmentation, and a platelet count of 7000 cells/mm 3 . On the second hospital day, she delivered a nonviable fetus. Continuous venovenous hemodialysis was initiated, and laboratory studies were notable for mildly prolonged coagulation times, hypofibrinogenemia, and d -dimer greater than 30 mg/L. A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) activity was 41% (normal, 70%–155%) and complement C3 and C4 were normal. Despite improving with supportive care, she developed seizures on hospital day 10. The C3 was now low at 70 mg/dL (normal, 79–152), and therapeutic plasma exchange (TPE) was initiated. Over the next 2 weeks, she received daily TPE with recovery of both platelets and renal function, and ADAMTS13 activity normalized. After discontinuation of TPE, however, platelets and renal function worsened, and ADAMTS13 activity fell to 19%. TPE was reinitiated, and rituximab was administered. Renal biopsy obtained 48 days after her presentation revealed focal cortical infarction, congested glomeruli with loss of capillary endothelial cells, mesangiolysis with embedded erythrocyte fragments, and intimal edema with endothelial activation in an interlobular artery. Immunofluorescence was notable for segmental granular mesangial immunoglobulin (Ig)A (sparse 1–2+), IgM (1+), C3 (1+), kappa (1–2+), and lambda (sparse trace – 1+). Segmental capillary wall staining for C3 (1–3+) was present, and electron microscopy revealed subendothelial widening, capillary loop collapse, and endothelial cell swelling with no electron dense deposits. Platelets and renal function normalized after 29 plasma exchanges. Relevant serologic testing for antinuclear antibodies, antiphospholipid antibody syndrome, and human immunodeficiency virus was negative. Nine months after her presentation, genetic testing identified a heterozygous pathogenic mutation in thrombomodulin ( THBD ; c.1456G>T; p.Asp486Tyr), and a previously unreported heterozygous mutation in complement factor H related protein-5 ( CFHR5 ; c.1357C>G; p.Pro453Ala) of unknown significance, consistent with a diagnosis of atypical hemolytic uremic syndrome. Two years later she remains in remission with serum creatinine 0.7 mg/dL (estimated glomerular filtration rate, >60 mL/min/1.73 m 2 ), normal urine protein excretion, and normal hematologic parameters.
Classification of thrombotic microangiopathy
This case demonstrates the complexity of thrombotic microangiopathy (TMA) syndromes and the challenges clinicians face in arriving at a diagnosis and delivering targeted therapy. TMAs are clinical syndromes defined acutely by the presence of fragmented red cells, anemia, thrombocytopenia (<150,000 cells/mm 3 ), and microvascular thrombi, with end-organ dysfunction attributed to small vessel occlusion. Chronic, smoldering TMA, particularly renal limited, may not exhibit significant anemia and thrombocytopenia. Clinically, TMA is most commonly represented by thrombotic thrombocytopenic purpura (TTP), enteric infection-associated hemolytic uremic syndrome (HUS), complement-mediated atypical HUS (aHUS), and disseminated intravascular coagulation. Many clinical syndromes, however, are characterized by 1 or more of these features, including this patient in whom disseminated intravascular coagulation secondary to fetal demise was initially considered.
Moschcowitz’ 1924 report of a 16-year-old with TTP was followed decades later by Gasser’s description of HUS in a group of children following diarrheal illness. The link between enteric infection by Escherichia coli and HUS was established in the 1980s, and molecular diagnostics have firmly established the role of complement dysregulation in the pathogenesis of aHUS. Classification rubrics, however, are challenging, given the large number of conditions associated with TMA, the inconsistent use of terminology, and the recognition of overlapping features in these diseases. Many would agree that TTP is characterized by deficiency of ADAMTS13 (activity classically < 5%–10% in TTP), HUS by the presence of Shiga or Shiga-like toxin producing enteric infection (STEC-HUS), and aHUS by complement-mediated disease with the frequent identification of complement-related mutations. Nonetheless, the list of TMA-associated conditions is long ( Box 1 ).
A disintegrin and metalloproteinase with a thrombospondin type 1 motif, member 13 (ADAMTS13) deficiency
Thrombotic thrombocytopenic purpura
Infection associated
Shiga or Shiga-like toxin hemolytic uremic syndrome (HUS, typical HUS, Shiga-toxin–associated HUS)
Campylobacter jejuni
Streptococcus pneumonia
Human immunodeficiency virus
Cytomegalovirus
Epstein–Barr virus
Parvovirus B19
BK virus
Influenza
Abnormality in complement regulation
Atypical hemolytic uremic syndrome
Disseminated intravascular coagulation
Autoimmune/connective tissue disease
Systemic lupus erythematosus
Antiphospholipid antibody syndrome
Scleroderma
Vasculitis/glomerulonephritis
Antineutrophil cytoplasmic antibody-associated vasculitis
Membranous nephropathy
Immunoglobulin A nephropathy
Cryoglobulinemia
Pregnancy
Malignant hypertension
Drugs
P2Y 12 antagonists
Calcineurin inhibitors/sirolimus/everolimus
Quinine
Estrogen/progesterone
Gemcitabine/mitomycin C
Interferon
Vascular endothelial growth factor inhibitors/tyrosine kinase inhibitors
Cocaine
OPANA (R) ER
Metabolic/cell signaling
Cobalamin responsive methylmalonic acidemia
Diacylglycerolkinase epsilon mutation
Malignancy
Post organ transplant
Histologically, TMA may be indistinguishable among the different etiologies, with biopsy findings of endothelial injury and thrombus formation, and in the kidney, both intravascular and intraglomerular fibrin thrombi. In the acute phase of aHUS, vessels display intravascular fibrin thrombi and endothelial swelling and activation, leading to vascular obstruction. Chronically, the glomerular changes may evolve into a membranoproliferative-appearing pattern, although immunofluorescence is often negative for immunoglobulins and electron dense deposits are absent. Chronic vascular changes include vessel wall sclerosis and organizing thrombi, with subendothelial widening and deposition of flocculent material. In contrast, however, to aHUS histology, TTP is characterized by microvascular thrombi rich in platelets, fibrin, and von Willebrand factor (vWF) protein. TTP histology may not invoke the same degree of endothelial damage and edema as aHUS, although the histologic appearance of these 2 diseases often overlaps. The renal microvasculature is relatively spared in TTP, and although acute kidney injury is common in TTP, the creatinine is often less than 2.2 mg/dL and the need for renal replacement therapy occurs in less than 30% of patients. Despite the decline in ADAMTS13 activity in our patient, the activity greater than 10% and severity of TMA histology evident on biopsy were most supportive of aHUS.
Epidemiology
Diagnostic uncertainty has confounded epidemiologic, natural history, molecular, and therapeutic studies to this day. The true incidence of TMA is unknown, although 2005 data from the Oklahoma TTP-HUS Registry provide a standardized estimate of suspected TTP-HUS of 11.29 per million per year, with a standardized incidence rate of 1.78 from the most recent 2013 data for confirmed ADAMTS13 deficient TTP. Non–diarrheal-associated HUS (presumably aHUS) in children has been estimated at 2 per million. Clinical experience suggests that the diagnosis of TMA, and aHUS in particular, has often been missed. Patients with TMA syndromes typically present during an acute phase requiring hospitalization, and thus hospital-based registries are reasonable sources to capture new cases. Fig. 1 shows the distribution of TMA from the last 24 adult inpatients recruited in 2014 to the Johns Hopkins TMA Registry. The case under discussion is not included in these data. In this series, classical TTP (defined by a low ADATMS13 [<15%] at initial presentation) comprised more than one-half of TMA, whereas STEC-HUS or typical HUS comprised the minority. Owing to logistical reasons, posttransplant TMA was underrecruited. In our series, one-half of the TTP cases were relapsed episodes; therefore, non-TTP TMA outnumbered new TTP 2 times to 1, a statistic that should inform clinicians considering TMA diagnostics. Originally considered a disease of childhood, 40% to 60% of aHUS actually presents after the age of 18, with only 50% of carriers developing disease by age 45. Female predominance is present in TTP and aHUS, representing more than 50% to 75% of cases ( Table 1 ). Infection is a common trigger for aHUS, and in women, pregnancy associates with disease in 20% ( Table 2 ). TTP most commonly presents in the second and third trimesters, in part facilitated by the physiologic decline in ADAMTS13 activity with gestation. With aHUS, however, 79% occurred in the postpartum period in the largest series to date. The clinical phenotypes of TTP and aHUS are otherwise frequent mimics, and it is well-recognized that neurologic symptoms are as common in both. Female predominance in both TTP and non-TTP TMA has not been explained adequately, although many of the triggers associated with TMA—autoimmunity, collagen/vascular disorders, and pregnancy—are gender specific. A high prevalence of hypertension and diabetes is noted in all TMA (see Table 2 ), and in this series hypertension preceded the diagnosis of TMA in the majority of cases. Comorbidities may drive TMA or act as risk factors for the development of TMA, and in many patients, it is unclear as to the degree of their causal relationship (see Table 2 ). In the 11 non-TTP, non–STEC-HUS patients evaluated, only 3 were considered to be truly sporadic, whereas 8 were associated with diseases or clinical contexts in which TMA commonly occurs; hence, the classification of “aHUS” (3 patients) and “Other TMA” (8 patients) adopted in the Tables and Figures. TMA is a multifactorial disease, and depends on the relative strengths of constitutional predisposition and external triggers. Strong triggers, such as toxin-associated colitis, may drive HUS in individuals with minimal or no genetic risks, whereas weaker triggers, such as calcineurin inhibitor exposure, may drive disease in those with strong predisposing risks.
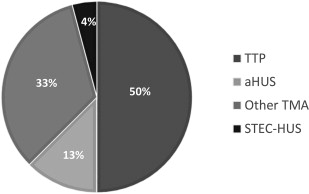
| N | Age (Range) | Female | Black Race | Relapse | |
|---|---|---|---|---|---|
| TTP | 12 | 39 (26–64) | 5/12 | 11/12 | 6/12 |
| aHUS | 3 | 53 (46–62) | 2/3 | 0/3 | 0/3 |
| Other TMA | 8 | 48 (24–69) | 6/8 | 5/8 | 0/8 |
| STEC-HUS | 1 | 84 | 1/1 | 0/1 | 0/1 |
| Total | 24 | 46 (24–84) | 14/24 | 16/24 | 6/24 |
| HTN/DM | Collagen Vascular | Cancer | Transplant | Drug | HIV | Pregnancy | Diarrhea | |
|---|---|---|---|---|---|---|---|---|
| TTP | 6/12 | 0 | 0 | 0 | 0 | 0 | 0 | 0 |
| aHUS | 3/3 | 0 | 0 | 0 | 0 | 0 | 0 | 1/3 |
| Other TMA | 4/8 | 2/8 | 2/8 | 2/8 renal, BMT | 2/8 quinine, tacrolimus | 1/8 | 1/8 | 0/11 |
| STEC-HUS | 1/1 | 1/1 | 0 | 0 | 0 | 0 | 0 | 1/1 |
| Total | 14/24 | 3/24 | 2/24 | 2/24 | 2/24 | 1/24 | 1/24 | 2/24 |
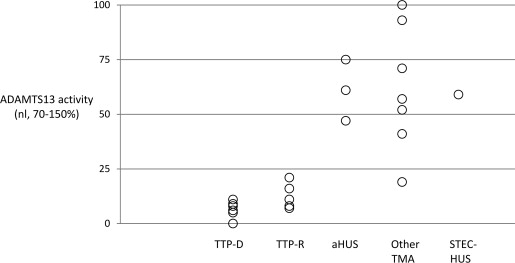
In our case, the adult age at presentation would historically have argued against a constitutional disorder of complement regulation, a feature now recognized to be incorrect. Moreover, the reduced ADAMTS13 level during her disease relapse is consistent, in our center’s experience, with the spectrum of disease attributable at least in part to ADAMTS13 pathology ( Fig. 2 ). Because her genetic diagnosis is recognized to be in the causal pathway for aHUS (vide infra), her illness can be recast as a strong predisposition to developing aHUS, ultimately triggered by the additional risk factor of pregnancy.
Complement pathway and regulation
Complement is a cascading network of proteins critical to innate immunity in defense against microorganisms, clearance of immune complexes, mediation of inflammation, and in embryonic and fetal development. Complement is regulated elegantly at multiple levels to minimize excessive activation and bystander injury. The early complement cascade is traditionally initiated via the classical, mannose-binding lectin, or alternative pathway, culminating in the formation of the C5 convertase with subsequent generation of the terminal complement complex C5b-9. C5b-9 forms a membrane-spanning pore capable of inducing sublethal and lethal cellular injury when regulatory control is absent. The prototypical example of C5b-9–mediated lethality of autologous cells is paroxysmal nocturnal hemoglobinuria, characterized by absent cell surface expression of complement regulators CD55 and CD59 owing to loss of their tethering glycophosphatidylinositol anchor. Nontraditional mechanisms for complement activation are now recognized, including thrombin-mediated cleavage of C5 to generate C5b-9 independent of early complement activation. Although all pathways of complement activation have the potential to injure tissue via generation of inflammatory anaphylatoxins C3a and C5a in addition to C5b-9, it is dysregulation of the alternative pathway that has received the greatest attention in the development of aHUS. Classical and mannose-binding lectin–based pathways were believed to likely be of particular relevance to other TMAs, such as seen in systemic lupus erythematosus, antiphospholipid antibody syndrome, and antibody-mediated allograft rejection. Nonetheless, recent data suggest that approximately 82% of kidney biopsies from a small series of aHUS patients contain immunohistochemical evidence of classical and/or mannose-binding lectin activation. These mechanisms of TMA and their particular relevance to aHUS, however, remain inadequately studied.
Initiation of the alternative pathway begins with hydrolysis—either spontaneous or triggered by contact with biological surfaces—of soluble phase C3 to C3(H 2 O). Hydrolyzed C3 can bind to factor B (CFB), which in the presence of factor D, is cleaved to C3(H 2 O)Bb. This soluble phase C3 convertase is stabilized and amplified by properdin, accelerating cleavage of additional C3 to C3b and the anaphylatoxin C3a. The C3 convertase is under negative regulation by factors H (CFH) and I (CFI). Binding of CFH to C3b prevents CFB binding, and also serves as a substrate for CFI-mediated cleavage and inactivation of C3b to iC3b. C3(H 2 O) has a slightly higher affinity for CFB than for CFH, favoring formation over degradation of the soluble C3 convertase. There exists a superfamily of Complement Factor H Related Proteins (CFHR1-5) located downstream of CFH in the Regulation of Complement Activation gene cluster on chromosome 1q32. The specific roles of CFHR proteins in complement regulation are loosely understood, but in part may serve as competitive inhibitors of CFH binding. Homozygous deletion of the gene region encompassing CFHR3 and CFHR1 is associated with the development of autoantibodies to CFH and the development of aHUS. The loss of CFHR1 may be the critical gene region. Activated C3b covalently links to the cell surface, where it can form surface-bound C3 convertase (C3bBb). Additional C3b binding to the C3 convertase forms the surface bound C5 convertase C3bBbC3b, which is capable of cleaving C5 to soluble C5a and surface-bound C5b. This leads to formation of the pore-forming membrane attack complex (MAC; C5b-9) via assembly of C5b with C6–C9. Some fraction of MAC may be liberated into the soluble phase (sMAC).
Additional surface phase regulation of the complement cascade is provided by cell surface-expressed complement receptor 1 (CR1), decay accelerating factor (DAF; CD55), membrane cofactor protein (MCP; CD46), thrombomodulin (THBD), and CD59. CR1 binds C3b and C4b (classical pathway), thereby facilitating clearance of opsonized particles as well as abrogating activity of the C3/C5 convertases. Both DAF and MCP facilitate C3/C5 convertase disassembly. THBD—mutated in the case under discussion—is an endothelial surface protein that facilitates thrombin activation of activated protein C, inhibits thrombin cleavage of C5, promotes activation of thrombin-activatable fibrinolysis inhibitor (TAFI; carboxypeptidase B) to inactivate C3a and C5a, and facilitates CFI-mediated inhibition of C3b. CD59 antagonizes C9 insertion into C5b-8. In conjunction with soluble vitronectin and clusterin, which also impair C5b-9 assembly, a final opportunity for cell protection against MAC-induced cytotoxicity is provided. Notably, some coagulation factors may function to downregulate complement as well, with plasminogen (PLG) binding C3, C3b, and C5. PLG enhances CFI activity, and activated plasmin is capable of cleaving C5 and C3b at sites distinct from CFI.
Maintenance of endothelial health
It is recognized increasingly that the maintenance of endothelial health, as highlighted by the case under discussion, lays at the intersection of complement, coagulation, cell-mediated immunity, and state of endothelial activation. TMA does not develop in the context of a single stimulus (see Box 1 , Tables 1 and 2 ). Endothelial injury and activation occur through a variety of mechanisms, including trauma, ischemia–reperfusion injury, drug-induced toxicity, infection, shear stress, free heme, complement activation, and inborn errors of cellular metabolism and signaling. Rather than progressing in an orderly, linear fashion, the pathogenesis of TMA incorporates multiple intersecting, simultaneous, and ultimately amplifying pathways.
The predilection of TMA, and particularly complement-mediated injury, for the kidney remains unanswered. The renal microcirculation is distinct from most other vascular beds by virtue of a unique glycocalyx structure and fenestrated endothelium. Glomerular endothelial cells are critically dependent on vascular endothelial growth factor secreted by podocytes, with loss of such signaling (eg, administration of vascular endothelial growth factor antagonists, injury to the glomerular filtration barrier) associated with development of TMA. Moreover, podocytes are a source for complement components, potentially providing another mechanism for amplification of complement-mediated injury during activated states. Endothelial activation upregulates surface expression of vWF, tissue factor, and adhesion molecules such as P-selectin and intercellular adhesion molecule. Complement activation on the surface of platelets and activated endothelium generates the potent anaphylatoxins C3a and C5a, resulting in neutrophil recruitment and activation. Activated neutrophils adhere to the endothelium and migrate into tissue, resulting in further injury as well as disruption of basement membrane integrity through secretion of mediators such as tumor necrosis factor-α. Complement-stimulated neutrophils also increase production of prothrombotic tissue factor. THBD transcription and expression is downregulated in endothelial cells under many of these same stimuli, favoring complement activation and coagulation. These changes to the endothelium promote platelet adhesion and generation of microthrombi. The resultant hemolysis and liberation of heme directly activates C3 in the soluble phase, in addition to decreasing expression of surface phase regulators CD46 and CD55. In a potentially protective manner, activated platelets release CFH. Impaired CFH activity, however, as a consequence of mutation or inhibitory autoantibody, may activate platelets. Moreover, the endothelial expression of vWF from intracellular Weibel–Palade bodies is accompanied by secretion of co-localized CFH. Although increased vWF expression increases platelet adhesion, vWF-bound CFH can augment CFI-mediated complement inhibition. This complement-regulating pathway is lost with deficient CFH activity, thereby increasing both platelet adhesion and complement activation. Endothelial injury may also associate with decreased ADAMTS13 activity, because a large fraction of the ADAMTS13 enzyme is bound to endothelium, reinforcing the connection between endothelial injury, coagulation, and platelet activation (see Fig. 2 ; Table 3 ).
Related posts:
 Complement in Health and Disease
Complement in Health and Disease
 Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
Ultralarge Von Willebrand Factor–Induced Platelet Clumping and Activation of the Alternative Complement Pathway in Thrombotic Thrombocytopenic Purpura and the Hemolytic-Uremic Syndromes
 Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Paroxysmal Nocturnal Hemoglobinuria
Warm Autoimmune Hemolytic Anemia
Cold Agglutinin-Mediated Autoimmune Hemolytic Anemia
Current and Future Pharmacologic Complement Inhibitors
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


