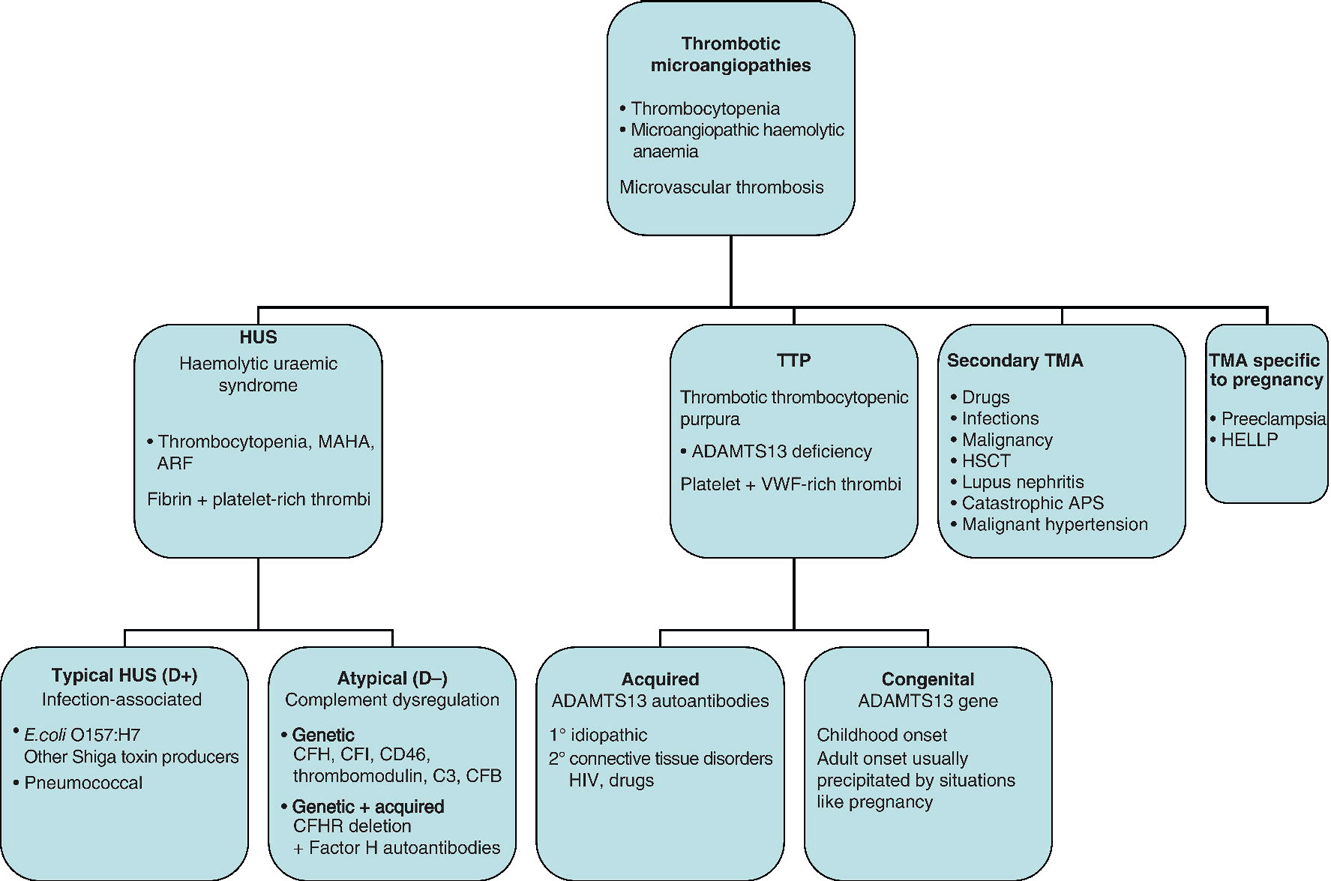
Chapter 11 Mari Thomas1 and Marie Scully2 1Haemostasis Research Unit, University College London, London, UK Thrombotic microangiopathies (TMAs) represent a spectrum of disorders characterized by thrombosis in small vessels. Thrombocytopenia is usually present, and review of the blood film confirms the microangiopathic process, with red cell fragmentation and often polychromasia. Considerable developments have been made in the field of TMA in recent years, and new genetic and autoimmune causes have been identified for the two major forms: haemolytic uraemic syndrome (HUS) and thrombotic thrombocytopenic purpura (TTP). However, the differential diagnosis of TMA may be difficult and remains a clinical one in the first instance. Differential diagnoses include disseminated intravascular coagulation (DIC); autoimmune disease e.g. lupus; severe hypertension; infections such as endocarditis; catastrophic antiphospholipid syndrome (CAPS); and malignancy (Figure 11.1). Figure 11.1 Subtypes of thrombotic microangiopathies (TMA). MAHA, microangiopathic haemolytic anaemia; ARF, acute renal failure; VWF, von Willebrand factor; APS, antiphospholipid syndrome; HELLP, haemolysis, elevated liver enzymes and low platelets; CFH, complement factor H; CFI, complement factor I; CFB, complement factor B; CFHR, complement factor H-related protein. Thrombotic thrombocytopenic purpura is a rare, acute, life-threatening disorder with an incidence of approximately six cases per million per year. There is no age limit, but TTP typically affects females between 30 and 40 years. TTP is characterized by thrombocytopenia, microangiopathic haemolytic anaemia (MAHA) and clinical features related to microvascular thrombosis. It occurs as a result of deficiency of the VWF-cleaving protease ADAMTS13, leading to the accumulation of ultralarge VWF multimers (ULVWF), increased platelet adhesion and aggregation and microthrombus formation. Most cases are acquired idiopathic, antibody-mediated TTP, but TTP may be precipitated by HIV infection, pregnancy, connective tissue disorders, pancreatitis or certain drugs [1]. In congenital TTP, ADAMTS13 deficiency results from recessive bi-allelic mutations in the ADAMTS13 gene. Thrombotic thrombocytopenic purpura is a clinical diagnosis and must be considered in any patient with MAHA and low platelet counts in the absence of another cause. Clinical features are variable, and the classical pentad of MAHA, thrombocytopenia, fever, neurological involvement and renal failure is rarely seen. In a recent study, 15 out of 40 patients required ITU admission, and 15% of cases were intubated and ventilated [2]. Neurological impairment is present in up to 80% of cases and may be fluctuating. Neurological features include headache, behavioural changes, transient ischaemic attacks, seizures or coma. Renal involvement is variable, resulting in reversible impairment. Cardiac features may include infarction, arrhythmias and sudden cardiac arrest. Elevated troponin-T levels at presentation are an accurate predictor of disease severity [3]. Gastrointestinal features occur in a third of cases [1]. Patients may present with symptoms of thrombocytopenia such as bruising and petechiae. Autopsy studies confirm multi-organ involvement in TTP, with platelet and VWF-rich microthrombi (in contrast to the fibrin deposits seen in HUS or an inflammatory component in DIC or autoimmune disease). The blood film shows red cell fragmentation, polychromasia, anaemia and low platelets (below 150 × 109 but usually <50 × 109). The reticulocyte count is increased and bilirubin is raised due to haemolysis. The clotting screen is typically normal. Lactate dehydrogenase (LDH) is disproportionately increased relative to the degree of haemolysis, due to associated tissue ischaemia. Virology screen pretreatment is necessary to exclude HIV-associated TTP and prior to therapy with large volumes of plasma. Prompt diagnosis and treatment is the key to survival in TTP. Without therapy, mortality is 90% – reduced with plasma exchange (PEX) to 15–20%. Plasma is a source of the missing/dysfunctional enzyme ADAMTS13, and PEX allows delivery of large volumes of plasma and assists in the removal of ULVWF and anti-ADAMTS13 antibodies in the case of acquired TTP. Plasma exchange must be started as soon as the diagnosis of TTP is suspected. If not, high-dose plasma infusions should be started as a holding measure, e.g. while awaiting transfer to a tertiary referral centre. Intensive PEX regimes are required in severe cases such as those with new/progressive neurological or cardiac symptoms. PEX should continue for at least 2 days after normalization of the platelet count. The UK guidelines recommend the use of solvent–detergent-treated FFP (Octaplas, Octapharma) to minimize pathogen exposure [4]. Complications of PEX include those related to the central venous catheter such as sepsis and reactions to plasma. As the majority of TTP cases are acquired antibody-mediated, immunosuppression is used to attain complete remission and/or prevent relapse. Steroids such as pulsed intravenous methylprednisolone or high-dose oral therapy are widely used in the treatment of TTP. Rituximab, a monoclonal anti-CD20 antibody, acting on B lymphocytes, results in depletion of antibody-producing B cells. Initially used in patients’ refractory to first-line therapy or with relapse, its use during acute presentation has also been demonstrated [2]. The first dose may be given over a longer period, e.g. 6–8 h, to minimize allergic-type symptoms although subsequent doses can be infused more quickly. Typical rituximab dosing is 375 mg/m2 weekly, although in patients undergoing concomitant PEX, it may be given every 3–4 days. Vincristine and cyclophosphamide (both pulsed and daily dosing) have been used with some success, although the reported numbers are extremely low. The use of ciclosporin has led to high rates of clinical and haematological response, but relapses are documented after cessation of therapy. Red cell transfusion may be required for rapid haemolysis, and all patients should receive folic acid supplementation. Platelet transfusions are contraindicated unless there is life-threatening haemorrhage as they are associated with disease exacerbation. Occult infection may prevent response to PEX or precipitate early relapse. An underlying infection should be actively sought. Low-dose aspirin (75 mg daily) and thromboprophylaxis are started with platelet count greater than 50 × 109/L and graduated elastic compression stockings used from admission.
Thrombotic Microangiopathies
2Department of Haematology, University College Hospital London, London, UK
Introduction
Thrombotic thrombocytopenic purpura
Clinical features of TTP
Laboratory features in TTP
Management of TTP
Plasma exchange (PEX)
Immunosuppression
Other immunosuppressive therapies
Supportive therapy
Role of ADAMTS13 assays
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


