Fig. 5.1
Virchow’s triad. Virchow’s triad describes the three main factors that predispose thrombosis
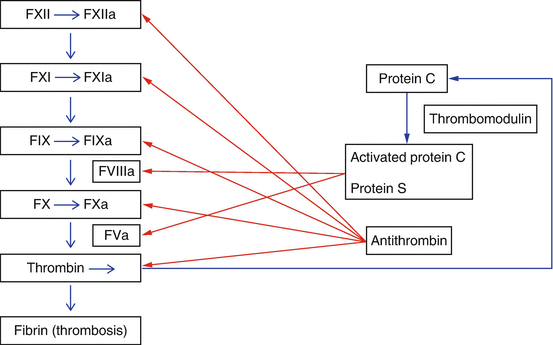
Fig. 5.2
Coagulation cascade. The intrinsic pathway of the coagulation cascade includes the most common inherited thrombophilias
Factor V Leiden (FVL), the most common inherited thrombophilia, is prevalent in 4–7 % of the Caucasian American population, 2 % Hispanic-Americans and 1 % African-Americans [2]. It is rare in native African and Asian populations [3]. FVL is characterized by a poor anticoagulant response to activated protein C (APC) . APC is a natural anticoagulant that degrades activated factors V and VIII (FVa and FVIIIa). The FVL mutation is due to a single point mutation in the factor V gene affecting the first APC cleavage site of FVa, where arginine is replaced by glutamine at position 506. This mutated cleavage site is less susceptible to proteolytic inactivation by APC, known as APC resistance (APCR). Less common causes of APCR include other rare factor V mutations, antiphospholipid antibodies, increased estrogen states, and cancers. APCR is detected by functional activated partial thromboplastin time-based coagulation assays using factor V-deficient plasma. The FVL gene is confirmed by genetic polymerase chain reaction (PCR) testing.
Prothrombin G20210A mutation (PG) is the second most common inherited thrombophilia, occurring in 2 % of the Caucasian population and is rare in non-Caucasians [4]. It results from a substitution of guanine for adenine at nucleotide 20210 in the prothrombin gene leading to increased prothrombin plasma levels. The mutant prothrombin gene is diagnosed by genetic testing.
Protein C (PC) and protein S (PS) are vitamin K-dependent glycoproteins. APC and its cofactor PS inhibit FVa and FVIIIa and suppress thrombin generation. PS additionally inhibits the prothrombinase and tenase complexes. PC deficiency affects 0.2–0.4 % of the general population [4] and PS deficiency 0.16–0.21 % [5]. Acquired protein C and S deficiencies occur in disseminated intravascular coagulation (DIC), liver disease, and with the use of vitamin K antagonist therapy (usually warfarin). Increased estrogen states, such as pregnancy or oral contraceptive use, may also decrease Protein S levels. Functional assays are used to detect Protein C and S deficiencies and immunoassays detect PC, free and total PS antigen levels.
Antithrombin (AT) is a natural anticoagulant that inhibits thrombin and activated factors IX, X XI, and XII. AT inhibition is markedly enhanced 1000-fold by heparin. AT deficiency has a prevalence of 0.02 % in the general population [4, 6]. Functional assays and immunoassays are available to detect AT deficiency. Acquired AT deficiency is seen in DIC, liver disease, nephrotic syndrome, surgery, asparaginase therapy, and heparin use.
Thrombophilia testing is recommended in patients suspected of having an inherited thrombophilia: unexplained thrombosis at young age (<50 years), recurrent thrombosis, family history of thrombosis, and thrombosis at unusual locations. Timing of testing is important as various factors can result in misleading results, for example anticoagulant therapy or acute thrombosis may lower PS and AT levels. Ideally, testing should be performed once off anticoagulation therapy for at least 2–3 weeks at the completion of the initial 3 month anticoagulation period. The aim of testing is to detect individuals at increased risk for recurrent disease and to determine if extended anticoagulation is required. Any abnormal results should be confirmed on repeat testing using an independent blood sample.
Individuals with any inherited thrombophilia and acute pulmonary embolism are treated with standard therapeutic anticoagulation for a minimum of 3 months. Precautions for anticoagulation include overlapping parenteral anticoagulant for at least 5 days with initial vitamin K antagonist therapy to prevent warfarin-induced skin necrosis in PC and PS deficiency and AT replacement may be required to achieve adequate anticoagulation with heparin therapy in AT deficiency. Individuals found to have more than one inherited thrombophilia, homozygous FVL or PG, or PC, PS, or AT deficiency are more prone to recurrent disease (than heterozygous FVL or PG) and should be considered for long-term anticoagulation.
Acquired Thrombophilic States and Thromboembolism
Paroxysmal Nocturnal Hemoglobinuria
Paroxysmal nocturnal hemoglobinuria (PNH) is a rare acquired clonal stem cell disorder characterized by intravascular hemolysis, thrombosis, and bone marrow failure. PNH affects 1–5 cases per million population [7] and is usually seen in young adults (median age 42 years) of both genders, but may affect all ages. The median survival is 10–15 years, with a French group reporting median survival of 22 years [8]. Spontaneous remission occurs in about 10–15 % of patients at any time [9].
PNH results from an acquired somatic mutation in the X-linked phosphatidylinositol glycan-class A (PIGA), resulting in deficiency of surface proteins attached by the glycosylphosphatidylinositol (GPI) anchor. More than 180 PIGA gene mutations have been identified in patients with PNH. Both CD55 and CD59 are among the missing GPI-anchored proteins. CD55 regulates the formation and stability of the C3 and C5 convertases and CD59 prevents the formation of the membrane attack complex (MAC). Complement activation occurs through a series of reactions that leads to formation of the MAC, which results in lysis of the target cell. Absent complement regulatory proteins on PNH erythrocytes accounts for terminal complement mediated hemolysis.
PNH results in variety of clinical features, ranging from asymptomatic disease to life-threatening thrombosis (Table 5.1).
Table 5.1
Clinical manifestations of PNH
Intravascular hemolysis | Thrombosis | Bone marrow failure |
|---|---|---|
Anemia Fatigue Shortness of breath Hemoglobinuria Renal failure Abdominal pain Back pain Chest pain Erectile dysfunction Esophagospasm Dysphagia Cholelithiasis Jaundice | Deep venous thrombosis Pulmonary emboli Intra-abdominal: hepatic, splenic, mesenteric, and renal vein thrombosis Cerebral vein thrombosis Retinal vein thrombosis Dermal vein thrombosis Arterial thrombosis (less common) | Anemia Neutropenia-infections Thrombocytopenia—bleeding Aplastic anemia Myelodysplastic syndrome Progression to AML (rare) |
The severity of hemolysis is related to the size of the PNH cell clonal population. Complications related to the presence of free hemoglobin in the plasma include renal toxicity and nitric oxide depletion. Nitric oxide regulates smooth muscle tone and its depletion leads to smooth muscle contraction, including vasoconstriction, contraction of gastrointestinal tract smooth muscle and pulmonary hypertension. Patients with a hemolytic crisis present with fatigue, dyspnea, chest pain, abdominal pain, esophageal spasms, dysphagia, back pain, erectile dysfunction, and renal failure. Most patients with PNH have some degree of bone marrow failure, even if there are no cytopenias present. Other bone marrow disorders may present with PNH, such as aplastic anemia (AA) or myelodysplastic syndrome (MDS), and are usually associated with a hypocellular marrow, pancytopenia, less intravascular hemolysis, and a smaller PNH clone.
Hemolysis is possibly an important contributing factor to the development of thrombosis as an increased incidence of thrombosis is seen in patients with larger PNH clones and thrombosis frequently follows a hemolytic episode [10]. The mechanism of thrombosis in PNH is not well understood but is possibly related to platelet activation initiated by hemolysis and complement, and increased procoagulant and fibrinolytic activity. Approximately 40 % of patients develop thrombosis, mostly venous, especially in the hepatic, portal, splenic, or cerebral veins [11]. The leading cause of death in PNH patients is thrombosis and the initial thrombotic event increases the relative risk of death in PNH five to tenfold [12].
Laboratory findings consistent with hemolytic anemia include increased reticulocyte count and LDH, decreased haptoglobin, hemoglobinuria, and hemosiderinuria, in addition to a negative direct Coombs test. Iron deficiency may result from urine-related iron losses. A bone marrow biopsy is not essential but may be helpful in evaluating patients with pancytopenia or suspected bone marrow disorders. The marrow may appear hypocellular, normocellular, or hypercellular, and erythroid hyperplasia is common. Appropriate imaging studies, such as abdominal ultrasound or magnetic resonance imaging, are obtained to evaluate any worrisome symptoms or signs.
Historically, the Ham test was used to diagnose PNH, where increased red cell lysis after exposure to acidified serum denoted a positive result. Low sensitivity and specificity were major limitations of this study, as well as the inability to determine the size of the PNH clone. Flow cytometry of peripheral blood is the preferred diagnostic test. Flow cytometry detects reduced levels of GPI-anchored proteins (CD55 and CD59), and measures the presence and size of the PNH clonal population. PNH type III cells express complete deficiency of GPI-linked proteins, PNH type II cells show partial deficiency, and PNH type I cells have normal protein levels.
Patients with PNH are classified into clinical subcategories : (1) classic PNH (intravascular hemolysis without another bone marrow abnormality, and usually a large clone >50 %), (2) PNH in the setting of other bone marrow failure disorders, such as AA/PNH or MDS/PNH, or (3) subclinical PNH (very small PNH clone without hemolysis).
Treatment depends on the severity of disease. Allogeneic hematopoietic cell transplantation and eculizumab are the only established therapies for treatment of classic PNH. Eculizumab is approved for treatment of PNH and is indicated in patients with significant hemolysis and symptomatic disease. Eculizumab is a humanized monoclonal antibody that blocks complement C5 and prevents formation of the membrane attack complex (Fig. 5.3). Eculizumab reduced hemolysis and thrombotic risk, and improved anemia, fatigue, and quality of life in PNH patients [10]. Treatment with eculizumab is continued indefinitely as it does not impact the underlying stem cell disorder or any associated BM failure syndromes. Eculizumab increases the risk of Neisseria meningitidis with chronic use but is otherwise well tolerated.
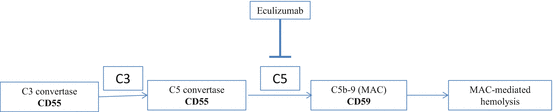
Fig. 5.3
PNH complement activation. CD55- and CD59-deficient red cells in PNH are lysed by activated complement. Complement C5 is inhibited by the drug eculizumab
Allogeneic hematopoietic cell transplantation (HCT) is the only cure for PNH. However, HCT is associated with significant morbidity and mortality compared to the relatively safe eculizumab. The decision for HCT should be made by physicians experienced in transplantation and PNH, and may be reserved for patients for whom eculizumab fails, or with associated severe aplastic anemia or high risk MDS.
Patients with associated BM failure disorders do not require PNH-specific therapy and treatment should be aimed at the underlying BM failure disorder. Asymptomatic patients (subclinical PNH) do not need any treatment for PNH. Other supportive therapies include red blood cell transfusions for severe anemia, and supplementation with iron for iron deficiency and folate for hemolysis. Prophylactic anticoagulation is not needed for patients without prior thrombosis. However, PNH patients treated with eculizumab and history of prior thrombosis should continue anticoagulation due to the increased risk of a recurrent event [13].
Heparin-Induced Thrombocytopenia
Heparin-induced thrombocytopenia (HIT) is a potentially life-threatening condition that occurs following exposure to heparin and affects approximately 1–5 % of patients on heparin. Venous and/or arterial thrombosis is the major clinical finding in HIT. Mortality rates up to 20 % were seen in patients with HIT, but have improved with early recognition and intervention.
Two forms of HIT are recognized. Type I HIT (nonimmune heparin-associated thrombocytopenia) is characterized by a mild, transient fall in platelet count (rarely <100,000/μL) that typically develops within the first 2 days of heparin exposure and resolves with continued heparin use. Type I HIT is not associated with thrombosis and is of no clinical significance. Type II HIT is immune-mediated and associated with thrombosis. Type II HIT will be discussed from hereon.
HIT is caused by IgG autoantibodies directed against complexes of platelet factor 4 (PF4) and heparin. Platelet Fc receptors bind the antibody-heparin-PF4 complex, resulting in activation of platelets and subsequent procoagulant response with thrombin generation. HIT antibodies also bind to endothelial cells, leading to endothelial cell activation with increased tissue factor and thrombin generation [14]. Thrombocytopenia occurs when the IgG-coated platelets are removed by the macrophages of the reticuloendothelial system and during platelet consumption for thrombi formation.
The majority of HIT patients (85–90 %) present with thrombocytopenia, less than 150,000/μL, 5–14 days after exposure to heparin, regardless of the dose or route of administration. HIT occurs predominantly with exposure to unfractionated heparin, and is more common in women and surgical patients [15, 16]. The platelet count falls greater than 50 % from baseline with a mean nadir of 60,000/μL. However, 5 % of patients may not develop thrombocytopenia but demonstrate a 30–50 % drop in platelet count. Platelet counts rarely fall below 20,000/μL and bleeding is hardly seen. Rapid onset HIT (within the first 24 h of exposure) occurs in patients previously exposed to heparin within the previous 100 days secondary to the presence of HIT antibodies already in the patient’s plasma. Delayed onset HIT has been described and is seen up to 3 weeks after heparin exposure, caused by high titers of platelet-activating heparin-induced IgG resulting in heparin-independent platelet activation, in addition to heparin-dependent activation [17].
Thrombosis is seen in up to 50 % of patients with HIT. Thrombocytopenia often precedes thrombosis. Venous thromboembolism occurs more frequently than arterial thrombi. Pulmonary embolism was the most common life-threatening thrombotic event, occurring in 25 % of all patients [18]. Complications of HIT include ischemic limb gangrene resulting in amputation, skin necrosis, myocardial infarction, stroke, organ infarction, or post-intravenous heparin anaphylaxis. The increased thrombotic risk persists for days to weeks after discontinuation of heparin [19].
The initial suspicion of HIT is clinical. A clinical scoring system, such as the 4T’s score, is used to guide the determination of HIT and need for further investigation. The 4T’s score has been validated and assesses the degree of Thrombocytopenia, Timing relative to heparin exposure, presence of Thrombosis, and other causes for thrombocytopenia. One study reported a negative predictive value of a low 4T’s score of 0.998 and positive predictive value of an intermediate and high score of 0.14 and 0.64, respectively [20, 21]. Essentially, a low 4T’s score rules out HIT and intermediate or high scores require further testing for HIT.
HIT antibodies can be demonstrated by immunoassays (enzyme-linked immunosorbent assays [ELISA]) that detect anti-PF4/heparin antibodies or functional assays (serotonin-release assay [SRA]) that detect heparin-dependent platelet activation by patient serum. It is important to note that all cases of HIT are caused by platelet-activating antibodies, but not all PF4/heparin complex antibodies result in HIT (some antibodies are clinically insignificant). There is data showing clinical correlation between immunoassays and SRAs. A weakly positive ELISA (OD 0.40 to <1.00) was associated with a less than 5 % low probability of a strongly positive SRA and increased to approximately 90 % with an OD greater than 2.00 [22]. The SRA is more specific and is considered the “gold standard” for diagnosing HIT, but it is less widely available, more complex and may take up to several days. The SRA is especially useful in cases of indeterminate immunoassays (OD 0.4 to 2.0).
Patients with HIT (presumptive due to intermediate or high 4T score, or confirmed by SRA) should have all heparin products stopped, including heparin flushes. A non-heparin anticoagulant should then be substituted due to the increased thrombotic risk, unless there is bleeding or a high risk of bleeding. Treatment should not be delayed while awaiting confirmative laboratory testing. Early intervention may prevent thrombotic events and subsequent morbidity and mortality [23]. Alternative anticoagulants used to treat HIT include direct thrombin inhibitors (argatroban, bivalirudin), fondaparinux (anti-factor Xa inhibitor), and danaparoid. The choice of anticoagulant agent depends on the patient’s renal and hepatic function. Vitamin K antagonists (VKA) such as warfarin should not be given in acute HIT and should only be started when the platelet count has recovered to at least 150,000/μL due to the increased risk of venous limb gangrene and skin necrosis [24]. The non-heparin anticoagulant should be continued for a minimum overlap of at least 5 days with VKAs and the target international normalized ratio (INR) has been reached. If the patient had been on VKA therapy at the time HIT was diagnosed, Vitamin K should be given. Target-specific oral anticoagulants, such as rivaroxaban and dabigatran, should be effective treatment for HIT based on principle, however, clinical data is limited and their off-label use is considered on a case by case basis [25]. Anticoagulation with a non-heparin anticoagulant should be continued for at least 2–3 months in patients with HIT, and for at least 3–6 months in patients with associated thrombosis. If the diagnosis of HIT is excluded, heparin may be resumed. Patients with confirmed HIT should avoid heparin, including low molecular weight heparin (LMWH) due to strong cross-reactivity of the HIT antibody, for life.
Antiphospholipid Syndrome
Antiphospholipid syndrome (APS) is an acquired thrombophilic state characterized by venous and arterial thrombosis, as well as recurrent miscarriages and pregnancy complications, in the presence of antiphospholipid antibodies [26]. Antiphospholipid antibodies (APL) are autoantibodies directed against phospholipids and phospholipid-binding proteins. APL may occur in the absence of disease (primary APS), or are often seen in association with other diseases, especially systemic lupus erythematosus. They occur in other rheumatologic conditions, or may be associated with infections—bacterial or viral, medications such as hydralazine and phenytoin, and malignancy. APL may be seen in healthy individuals, up to 5 %, but these are usually transient and of no clinical significance. Thrombosis appears to occur through various mechanisms including activation of platelets and endothelial cells, increased tissue factor and thrombin generation, and interference with antithrombotic pathways [27].
APS is diagnosed based on the revised Sapporo classification, which requires at least one clinical and one laboratory criteria [28]. Clinical criteria include either the confirmed presence of venous, arterial, or small vessel thrombosis in any tissue or organ, or pregnancy complications such as three or more unexplained early pregnancy losses, one or more unexplained fetal death of at least 10 weeks gestation, or one or more preterm delivery prior to 34 weeks of gestation due to eclampsia, preeclampsia, or placental insufficiency. The persistence of the following APL on two or more occasions at least 12 weeks apart must be demonstrated on laboratory testing: lupus anticoagulant (LA), anticardiolipin antibodies (ACL) immunoglobulin G (IgG) and immunoglobulin M (IgM) in moderate or high titers, or anti-β2-glycoprotein-I antibodies (anti-β2GPI) in moderate or high titers. Lupus anticoagulants are detected by various functional coagulation assays that result in prolonged clotting times. ACL and β2GPI antibodies are detected by ELISA. Triple positivity is associated with a higher risk of thrombosis. Testing for additional antibodies, such as antiphosphatidylserine antibodies, is not recommended as their association with thrombosis is not well established.
Venous thromboembolism is the most common clinical manifestation of APS. Other clinical findings that do not meet criteria include thrombocytopenia, livedo reticularis, valvular disease, neurological manifestations such as memory impairment and white matter lesions on MRI, and rare episodes of bleeding due to prothrombin antibodies. Catastrophic APS may be seen in a small proportion of patients who develop widespread thrombosis with multiorgan failure.
Patients with APS and venous thromboembolism are treated using standard anticoagulation with unfractionated heparin or LMWH, followed by warfarin targeting an INR of 2–3 [29]. LA may interfere with PTT and PT/INR anticoagulation assays and in such cases other studies are employed, such as anti-factor-Xa, chromogenic factor X, or factor II activity, to determine the level of anticoagulation. The recommendation is to continue anticoagulation indefinitely in patients with confirmed APS due to the high risk of recurrence [30]. Target specific oral anticoagulants are not recommended for treating VTE in APS until more evidence becomes available for their use in such patients. The treatment of arterial thrombosis is controversial and options include antiplatelet therapy, anticoagulant therapy, or both. Treatment for catastrophic APS includes anticoagulation and glucocorticoids, and possibly IVIG, plasma exchange, and other immunosuppressants such as cyclophosphamide [31]. Pregnant women with APS should be treated with LMWH and/or low-dose aspirin based on their history and clinical presentation.
Myeloproliferative Neoplasms
Myeloproliferative neoplasms (MPNs) are hematopoietic stem cell clonal disorders characterized by the overproduction of one or more of hematopoietic cells. MPNs have the potential to evolve into myelofibrosis or acute myeloid leukemia. The four most common MPNs are polycythemia vera (PV), essential thrombocythemia (ET), primary myelofibrosis (PMF), and chronic myeloid leukemia (CML). Other MPNs include chronic neutrophilic leukemia, chronic eosinophilic leukemia, and mastocytosis. CML is associated with a balanced translocation between 9 and 22 [t(9;22)9q34;11)] called the Philadelphia chromosome. PV, ET, and MF are Philadelphia chromosome negative and in many cases are characterized by JAK2 mutations. These mutations cause constitutive activation of the JAK2 tyrosine kinase (gain of function) resulting in increased cell proliferation and survival. Almost all patients with PV have a JAK2 V617F or exon 12 mutation, whereas ET and PMF display 50–60 % JAK2 V617F mutations [32]. Additional mutations, such as CALR and MPL, are detected in patients with JAK-2 negative MPNs.
MPNs are rare and seen in approximately 0.5–3 per 100,000 persons depending on the subtype [33]. MPNs, especially PV and ET , are associated with an increased risk of thrombosis, both venous and arterial. Thrombosis in unusual locations, such as the splanchnic circulation and cerebral veins, should raise the suspicion for an underlying MPN. MPNs accounted for 40 % of Budd Chiari syndrome and 31 % of portal vein thrombosis [34]. The pathogenesis of thrombosis is complex and involves various processes, such as increased inflammatory cytokines, leukocyte-derived proteases, and endothelial adhesion molecules, leading to a procoagulant environment. Patients may present with fatigue, fever, night sweats, pruritus, early satiety due to splenomegaly, erythromelalgia, headaches, vision changes, and bleeding due to acquired von Willebrand disease.
Treatment depends on the underlying disease and aims to prevent or minimize complications, especially thrombotic risk in ET and PV [35]. Low dose aspirin, with or without hydroxyurea (an antimetabolite), is recommended in patients at high thrombotic risk, those with prior thrombosis and age greater than 60 years [36]. Phlebotomy and/ or hydroxyurea are used to control hematocrit in PV. Platelet lowering agents, such as hydroxyurea or anagrelide, are used in ET patients at increased thrombotic risk. Low risk or asymptomatic patients with MPN may be observed. Supportive measures such as red blood cell or platelet transfusions are used for significant cytopenias. Hematopoietic cell transplantation is the only curative treatment for PMF but is associated with significant transplant-related mortality and so is reserved for specific patients. Ruxolitinib is a JAK1/JAK2 inhibitor approved for treatment of certain MPNs, for whom hydroxyurea has failed or those with high risk MF.
Acute Pulmonary Thromboembolic Disease
Introduction
Venous thromboembolism (VTE) includes deep vein thrombosis (DVT) and pulmonary embolism (PE). The exact incidence of VTE is unknown but it is estimated at 300,000–600,000 cases each year in the United States [37]. VTE is responsible for the hospitalization of 150,000–250,000 patients annually and 100,000–200,000 deaths each year in the United States [38]. PE is the third most frequent cardiovascular disease [39, 40] and the most common preventable cause of hospital death.
Acute PE is the most serious presentation of VTE . It is a potentially life-threatening disease and up to 25 % of cases present as sudden death. Acute PE spans a wide spectrum of clinical outcomes mainly based on the right ventricle’s (RV) capacity to tolerate strain. Increased RV afterload causes RV dilation that then incites cardiac ischemia, coronary hypoperfusion, and hypotension that can progress to cardiogenic shock.
Pathogenesis
The three mechanisms of Virchow’s triad , blood stasis, endothelial injury, and blood hypercoagulability, are the basis of thrombus development (Fig. 5.1). First, blood stasis plays a major role in the pathogenesis of VTE, as thrombosis usually starts in the deep veins of the lower extremities, where blood flow is decreased. Thrombi, however, may also originate from higher in the pelvic and renal veins, inferior vena cava, upper extremity veins, and right heart. PE occurs when thrombi break off, travel to the lung, and occlude the main pulmonary artery, lobar branches, or more distal smaller vessels in the periphery. However, not all PE originates from DVT, but may arise in the pulmonary vasculature, which have been termed de novo PE [41, 42]. Second, endothelial injury and vessel-wall damage, more predominant in arterial thrombosis, results in collagen and tissue factor exposure, followed by events leading to thrombus formation. Exposed collagen recruits circulating platelets, which are subsequently activated and become a major component of the developing thrombus. Tissue factor initiates the coagulation cascade and generates thrombin and fibrin (Fig. 5.2) [43]. Last, conditions that shift the balance between natural anticoagulant and procoagulant processes may result in hypercoagulable states and thrombus formation.
Risk Factors and Stratification
A list of predisposing risk factors for VTE is in Table 5.2. Pulmonary embolism is associated with genetic and acquired risk factors. However, VTE can also occur in the absence of these factors.
Table 5.2
Risk factors for venous thromboembolism (VTE)
Inherited | Acquired |
|---|---|
Factor V Leiden Prothrombin G20210A Protein C deficiency Protein S deficiency Antithrombin deficiency Dysfibrinogenemia Hypoplasminogenemia Factor XII deficiency Sickle cell disease | Immobilization Acute medical illness Trauma Major surgery, especially orthopedic Malignancy Pregnancy Estrogen-containing contraceptives or hormone replacement therapy Medications, such as Tamoxifen, Thalidomide Central venous catheters Congestive heart failure Chronic lung diseases, such as chronic obstructive pulmonary disease (COPD) Nephrotic syndrome Liver disease Obesity Inflammatory bowel disease Sepsis Myeloproliferative neoplasm
Related posts: Sickle Cell Disease and Acute Chest Syndrome: Mechanisms and Pathogenenesis Sickle Cell Disease and Acute Chest Syndrome: Mechanisms and Pathogenenesis
 Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation Acute Pulmonary Complications of Bone Marrow and Stem Cell Transplantation
 Acute Pulmonary Manifestations of Hematologic Malignancies Acute Pulmonary Manifestations of Hematologic Malignancies
 Storage Lesion: Evolving Concepts and Controversies Storage Lesion: Evolving Concepts and Controversies
 Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes Sickle Cell Disease and Acute Chest Syndrome: Epidemiology, Diagnosis, Management, Outcomes
 Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery Transfusion-Related Immunomodulation (TRIM): From Renal Allograft Survival to Postoperative Mortality in Cardiac Surgery
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
 Get Clinical Tree app for offline access
Get Clinical Tree app for offline access

|