Thrombocytopenia
![]()
PLATELET BIOLOGY
![]() Platelets are anucleate blood cells that participate in primary hemostasis, the formation of a platelet plug at sites of vascular injury.
Platelets are anucleate blood cells that participate in primary hemostasis, the formation of a platelet plug at sites of vascular injury.
![]() Platelets are produced from megakaryocytes, multinucleate hematopoietic cells located in the bone marrow. Cytokines such as thrombopoietin are necessary for normal platelet maturation and release.
Platelets are produced from megakaryocytes, multinucleate hematopoietic cells located in the bone marrow. Cytokines such as thrombopoietin are necessary for normal platelet maturation and release.
![]() Once released into the circulation, the average life span of a platelet is 7 to 10 days. Platelets are removed from circulation when they are activated and utilized at sites of vascular injury or as they become senescent.
Once released into the circulation, the average life span of a platelet is 7 to 10 days. Platelets are removed from circulation when they are activated and utilized at sites of vascular injury or as they become senescent.
![]() At any given time, up to one-third of the platelet mass is stored in the spleen, providing a reserve of platelets that may be released during periods of physiologic stress.
At any given time, up to one-third of the platelet mass is stored in the spleen, providing a reserve of platelets that may be released during periods of physiologic stress.
![]() The normal platelet concentration in the blood is 150,000 to 400,000/μL as measured in most hospital laboratories.
The normal platelet concentration in the blood is 150,000 to 400,000/μL as measured in most hospital laboratories.
![]()
ETIOLOGY AND CLINICAL FEATURES OF THROMBOCYTOPENIA
Thrombocytopenia may occur due to
![]() Decreased production of platelets
Decreased production of platelets
![]() Increased consumption of platelets
Increased consumption of platelets
![]() Increased sequestration of platelets
Increased sequestration of platelets
![]() Any combination of these mechanisms (Table 19.1)
Any combination of these mechanisms (Table 19.1)
Regardless of the cause of thrombocytopenia, “platelet-type” bleeding is typically mucocutaneous and is characterized by petechiae, ecchymoses, epistaxis, and gingival and conjunctival hemorrhages. Less commonly, severe thrombocytopenia may lead to gastrointestinal, genitourinary, or central nervous system bleeding.
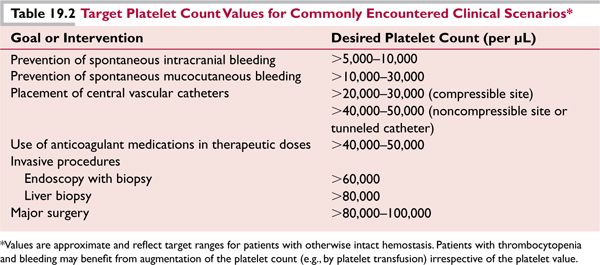
Spontaneous bleeding or bruising normally does not occur until the platelet count has fallen below 10,000 to 20,000/μL. The rate of decline of the platelet count may also influence the likelihood of unprovoked bleeding, presumably due to compensatory processes in remaining platelets that may occur over time with persistent thrombocytopenia. Patients with dysfunctional platelets may bleed with higher platelet counts. Patients with thrombocytopenia and platelet counts greater than 20,000 to 30,000/μL without bleeding usually do not require immediate treatment to increase the platelet count. A platelet count of 80,000 to 100,000/μL is generally adequate for hemostasis during most invasive procedures, including surgery (Table 19.2).
Disorders Characterized by Decreased Production of Platelets
Bone marrow failure syndromes
Congenital (amegakaryocytic thrombocytopenia, Fanconi anemia, dyskeratosis congenita, Schwachmann-Diamond syndrome, thrombocytopenia–absent radii syndrome,Wiskott-Aldrich syndrome) Acquired (aplastic anemia, amegakaryocytic thrombocytopenia)
Myelodysplasia
Marrow infiltration (neoplastic, infectious)
Chemotherapy-induced
Irradiation-induced
Cyclic thrombocytopenia (some cases)
Immune thrombocytopenia
Folate, B12, or iron (advanced cases) deficiency
Ethanolism
Disorders or Conditions Characterized by Increased Clearance of Platelets
Immune thrombocytopenia
Heparin-induced thrombocytopenia
Thrombotic thrombocytopenic purpura/hemolytic-uremic syndrome
Disseminated intravascular coagulation (HELLP syndrome)
Posttransfusion purpura
Neonatal alloimmune thrombocytopenia
Von Willebrand disease, type IIB
Cyclic thrombocytopenia (most cases)
Mechanical destruction (aortic valvular dysfunction; extracorporeal bypass)
Disorders Characterized by Increased Sequestration of Platelets
Hypersplenism (see Table 19.5)
Other Conditions
Artifactual (pseudothrombocytopenia)
Drug-induced (see Table 19.6)
Gestational thrombocytopenia
HIV-associated thrombocytopenia
Infection- and sepsis-related thrombocytopenia
Hemophagocytosis
Qualitative platelet disorder-related (Bernard-Soulier disease, gray platelet syndrome, May-Hegglin anomaly)
HIV, human immunodeficiency virus.
![]()
DISORDERS CHARACTERIZED BY DECREASED PRODUCTION OF PLATELETS
Bone Marrow Failure
![]() Congenital disorders, such as Fanconi anemia or dyskeratosis congenita, typically present early in life; these syndromes often cause depression of other blood cell lineages (i.e., white cells and red cells) in addition to the platelet count.
Congenital disorders, such as Fanconi anemia or dyskeratosis congenita, typically present early in life; these syndromes often cause depression of other blood cell lineages (i.e., white cells and red cells) in addition to the platelet count.
![]() Other congenital disorders such as congenital amegakaryocytic thrombocytopenia and the thrombocytopenia with absent radius (TAR) syndrome are characterized by isolated thrombocytopenia.
Other congenital disorders such as congenital amegakaryocytic thrombocytopenia and the thrombocytopenia with absent radius (TAR) syndrome are characterized by isolated thrombocytopenia.
![]() Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disorder featuring thrombocytopenia, eczema, and immunodeficiency. Thrombocytopenia may improve with splenectomy, but allogeneic hematopoietic stem cell transplantation alone is potentially curative for the disorder.
Wiskott-Aldrich syndrome (WAS) is an X-linked recessive disorder featuring thrombocytopenia, eczema, and immunodeficiency. Thrombocytopenia may improve with splenectomy, but allogeneic hematopoietic stem cell transplantation alone is potentially curative for the disorder.
![]() Adult patients with acquired amegakaryocytic thrombocytopenia initially may appear to have immune thrombocytopenia (ITP) (see ensuing section), but the bone marrow reveals markedly reduced or absent megakaryocytes. The disorder may progress to aplastic anemia.
Adult patients with acquired amegakaryocytic thrombocytopenia initially may appear to have immune thrombocytopenia (ITP) (see ensuing section), but the bone marrow reveals markedly reduced or absent megakaryocytes. The disorder may progress to aplastic anemia.
![]() Patients with acquired aplastic anemia rarely present with isolated thrombocytopenia. Marked bone marrow hypocellularity with decreased megakaryocytes would suggest this diagnosis (see Chapter 6).
Patients with acquired aplastic anemia rarely present with isolated thrombocytopenia. Marked bone marrow hypocellularity with decreased megakaryocytes would suggest this diagnosis (see Chapter 6).
Myelodysplasia
![]() Mild thrombocytopenia with macrocytosis, with or without anemia or neutropenia, in an older individual is a typical presentation of myelodysplasia (MDS). Isolated severe thrombocytopenia (less than 20,000/μL) without any other blood count abnormalities is not typical.
Mild thrombocytopenia with macrocytosis, with or without anemia or neutropenia, in an older individual is a typical presentation of myelodysplasia (MDS). Isolated severe thrombocytopenia (less than 20,000/μL) without any other blood count abnormalities is not typical.
![]() Bone marrow aspirate and blood smear may show megakaryocytic dysplasia (including small and mononuclear “micromegakaryocyte” forms) and maturation abnormalities of erythrocytic and granulocytic precursor cells. Concurrent cytogenetic abnormalities may be present (see Chapter 7).
Bone marrow aspirate and blood smear may show megakaryocytic dysplasia (including small and mononuclear “micromegakaryocyte” forms) and maturation abnormalities of erythrocytic and granulocytic precursor cells. Concurrent cytogenetic abnormalities may be present (see Chapter 7).
![]() For treatment of thrombocytopenia due to MDS, see Chapter 7. Note that thrombopoietin receptor agonists may be contraindicated in MDS due to a potential acceleration in transformation to acute leukemia.
For treatment of thrombocytopenia due to MDS, see Chapter 7. Note that thrombopoietin receptor agonists may be contraindicated in MDS due to a potential acceleration in transformation to acute leukemia.
Marrow Infiltration
Infiltration of the bone marrow by malignant cells may cause thrombocytopenia, but usually only after massive replacement of the marrow space by tumor cells or immature hematologic precursor cells has occurred. Examination of the bone marrow biopsy and aspirate is required.
![]() The acute and chronic leukemias, myeloma, and lymphoma are the most common tumors resulting in cytopenias due to neoplastic marrow infiltration and direct suppression of normal hematopoiesis with some tumor types.
The acute and chronic leukemias, myeloma, and lymphoma are the most common tumors resulting in cytopenias due to neoplastic marrow infiltration and direct suppression of normal hematopoiesis with some tumor types.
![]() Certain infections (such as tuberculosis and ehrlichiosis) can result in formation of granulomas in the bone marrow that supplant the normal marrow architecture.
Certain infections (such as tuberculosis and ehrlichiosis) can result in formation of granulomas in the bone marrow that supplant the normal marrow architecture.
![]() Effective treatment of the underlying condition should be expected to restore a low platelet count to the normal range, but platelet transfusions may be required initially if bleeding is present or invasive procedures are planned (Table 19.2).
Effective treatment of the underlying condition should be expected to restore a low platelet count to the normal range, but platelet transfusions may be required initially if bleeding is present or invasive procedures are planned (Table 19.2).
Irradiation and Chemotherapy
![]() Irradiation and/or myelotoxic chemotherapy induce thrombocytopenia via direct toxicity to megakaryocytes or more immature hematopoietic stem and progenitor cells. The degree and duration of thrombocytopenia depends on the intensity and the type of the myelotoxic regimen.
Irradiation and/or myelotoxic chemotherapy induce thrombocytopenia via direct toxicity to megakaryocytes or more immature hematopoietic stem and progenitor cells. The degree and duration of thrombocytopenia depends on the intensity and the type of the myelotoxic regimen.
![]() Chemotherapy-induced thrombocytopenia typically resolves more slowly than does neutropenia and/or anemia, especially following repetitive cycles of treatment.
Chemotherapy-induced thrombocytopenia typically resolves more slowly than does neutropenia and/or anemia, especially following repetitive cycles of treatment.
![]() Platelet transfusions may be given if required. Trials of novel platelet growth factors for thrombocytopenia due to specific chemotherapeutic regimens are ongoing.
Platelet transfusions may be given if required. Trials of novel platelet growth factors for thrombocytopenia due to specific chemotherapeutic regimens are ongoing.
This exceedingly rare disorder is characterized by episodes of thrombocytopenia that occur cyclically, typically every 3 to 6 weeks. The thrombocytopenia is frequently severe and may be associated with significant bleeding. Treatment with oral contraceptives (female patients), androgens, immunosuppressive agents (such as azathioprine), or thrombopoietic growth factor has led to responses in some cases.
Nutritional Deficiencies
Folate deficiency (commonly associated with alcoholism) and vitamin B12 deficiency may cause decreased megakaryocytopoiesis and thrombocytopenia, usually in conjunction with anemia. In contrast, thrombocytosis is typical in cases of significant iron deficiency; in very severe iron deficiency, however, thrombocytopenia may also occur. In any of these situations replacement of the deficient vitamin or mineral corrects the thrombocytopenia.
![]()
DISORDERS CHARACTERIZED BY INCREASED CLEARANCE OF PLATELETS
Immune Thrombocytopenia
ITP is an acquired autoimmune disorder of increased platelet destruction and decreased platelet production, causing thrombocytopenia that may lead to bleeding.
![]() Epidemiology. The annual incidence of ITP in adults has been estimated to be about 2 to 4 cases per 100,000 persons and increases with age. 1
Epidemiology. The annual incidence of ITP in adults has been estimated to be about 2 to 4 cases per 100,000 persons and increases with age. 1
![]() Pathophysiology.2 Pathogenic antiplatelet antibodies can be identified in approximately 75% of patients with ITP and are directed against the platelet glycoprotein complexes IIb/IIIa and/or Ib/IX. The antibody-coated platelets are cleared by reticuloendothelial macrophages in the liver and/or spleen, decreasing the platelet life span from approximately 7 days to less than 2 days. Platelet production also is impaired in ITP, possibly because of antiplatelet antibody binding to bone marrow megakaryocytes. Primary adult-onset ITP is generally idiopathic and becomes chronic, whereas secondary ITP occurs in association with disorders of lymphoproliferation (lymphoma or chronic lymphocytic leukemia (CLL)) or immune dysregulation (systemic lupus erythematosus, human immunodeficiency virus (HIV) infection).3 In contrast, ITP in children often follows a viral infection and frequently resolves spontaneously without specific therapy.
Pathophysiology.2 Pathogenic antiplatelet antibodies can be identified in approximately 75% of patients with ITP and are directed against the platelet glycoprotein complexes IIb/IIIa and/or Ib/IX. The antibody-coated platelets are cleared by reticuloendothelial macrophages in the liver and/or spleen, decreasing the platelet life span from approximately 7 days to less than 2 days. Platelet production also is impaired in ITP, possibly because of antiplatelet antibody binding to bone marrow megakaryocytes. Primary adult-onset ITP is generally idiopathic and becomes chronic, whereas secondary ITP occurs in association with disorders of lymphoproliferation (lymphoma or chronic lymphocytic leukemia (CLL)) or immune dysregulation (systemic lupus erythematosus, human immunodeficiency virus (HIV) infection).3 In contrast, ITP in children often follows a viral infection and frequently resolves spontaneously without specific therapy.
![]() Presentation. Typically, new-onset severe ITP (platelets <30,000/μL) manifests with petechial bruising and bleeding from mucous membranes, including conjunctival hemorrhages, gingival bleeding, and epistaxis. Milder disease (platelet count >50,000/μL) often presents as an asymptomatically low platelet count on routine blood work.
Presentation. Typically, new-onset severe ITP (platelets <30,000/μL) manifests with petechial bruising and bleeding from mucous membranes, including conjunctival hemorrhages, gingival bleeding, and epistaxis. Milder disease (platelet count >50,000/μL) often presents as an asymptomatically low platelet count on routine blood work.
![]() Diagnosis. ITP is a diagnosis of exclusion. New-onset, isolated thrombocytopenia with no other readily apparent cause (including medication-related) in an otherwise asymptomatic adult generally may be regarded as sufficient for the diagnosis of ITP and subsequent initiation of medical therapies (if appropriate based on the degree of thrombocytopenia; see below). 4
Diagnosis. ITP is a diagnosis of exclusion. New-onset, isolated thrombocytopenia with no other readily apparent cause (including medication-related) in an otherwise asymptomatic adult generally may be regarded as sufficient for the diagnosis of ITP and subsequent initiation of medical therapies (if appropriate based on the degree of thrombocytopenia; see below). 4
![]() The presence of other cytopenias, age greater than 60 years, or failure of primary therapy (corticosteroids for a trial of 1 week) should prompt bone marrow examination. The presence of abnormal or decreased numbers of megakaryocytes or abnormal marrow cellularity should redirect the diagnostic evaluation away from ITP.
The presence of other cytopenias, age greater than 60 years, or failure of primary therapy (corticosteroids for a trial of 1 week) should prompt bone marrow examination. The presence of abnormal or decreased numbers of megakaryocytes or abnormal marrow cellularity should redirect the diagnostic evaluation away from ITP.
![]() All patients should be screened for hepatitis B and hepatitis C virus (HBC/HCV) and HIV infection (see below) and undergo evaluation of the blood smear, direct antiglobulin test (DAT) (Coombs test), and blood type (for Rh status).
All patients should be screened for hepatitis B and hepatitis C virus (HBC/HCV) and HIV infection (see below) and undergo evaluation of the blood smear, direct antiglobulin test (DAT) (Coombs test), and blood type (for Rh status).
![]() Helicobacter pylori testing, antiphospholipid antibodies, and antinuclear antibodies may be useful in selected patients.
Helicobacter pylori testing, antiphospholipid antibodies, and antinuclear antibodies may be useful in selected patients.
![]() Treatment. Individuals with mild or moderate thrombocytopenia (platelets >30,000/μL) who do not require a higher platelet count for surgery or active bleeding should not receive treatment. Rather, they may be observed at regular intervals for disease progression. Adults with platelet counts of <20,000 to 30,000/μL or those with significant bleeding generally should be treated.4
Treatment. Individuals with mild or moderate thrombocytopenia (platelets >30,000/μL) who do not require a higher platelet count for surgery or active bleeding should not receive treatment. Rather, they may be observed at regular intervals for disease progression. Adults with platelet counts of <20,000 to 30,000/μL or those with significant bleeding generally should be treated.4
![]() Initial treatment (Table 19.3) generally consists of a short course of corticosteroids (prednisone 1 mg/kg/day for 7–10 days with subsequent rapid tapering or “pulse” dexamethasone cycles of 40 mg daily for 4 days). A significant increase in the platelet count should be seen within 3 to 7 days. In the event of a platelet response, prednisone may be tapered rapidly to a dose of 20 mg/day; thereafter, tapering should proceed more slowly (by dose decrements of no more than 5 mg/adjustment, no more frequently than once every 2 to 3 weeks). Dexamethasone cycles may be given every other week for four cycles or monthly for up to 6 months.
Initial treatment (Table 19.3) generally consists of a short course of corticosteroids (prednisone 1 mg/kg/day for 7–10 days with subsequent rapid tapering or “pulse” dexamethasone cycles of 40 mg daily for 4 days). A significant increase in the platelet count should be seen within 3 to 7 days. In the event of a platelet response, prednisone may be tapered rapidly to a dose of 20 mg/day; thereafter, tapering should proceed more slowly (by dose decrements of no more than 5 mg/adjustment, no more frequently than once every 2 to 3 weeks). Dexamethasone cycles may be given every other week for four cycles or monthly for up to 6 months.
![]() For patients with serious active bleeding and/or very severe thrombocytopenia (<5,000–10,000/ μL), intravenous immune globulin (IVIg; 1 gm/kg/day for 2 days) or anti-D (WinRho®, 75 μg/kg/ dose; appropriate for non-splenectomized, Rh blood type-positive, non-anemic patients only) can be administered in addition to corticosteroids in order to decrease clearance of antibody-coated platelets. Responses are generally seen within 3 to 5 days of IVIg or anti-D administration.
For patients with serious active bleeding and/or very severe thrombocytopenia (<5,000–10,000/ μL), intravenous immune globulin (IVIg; 1 gm/kg/day for 2 days) or anti-D (WinRho®, 75 μg/kg/ dose; appropriate for non-splenectomized, Rh blood type-positive, non-anemic patients only) can be administered in addition to corticosteroids in order to decrease clearance of antibody-coated platelets. Responses are generally seen within 3 to 5 days of IVIg or anti-D administration.
![]() Platelet transfusions may be administered if the presentation is complicated by serious (intracranial) bleeding. Transfused platelets are expected to be cleared very rapidly in the presence of antiplatelet antibodies, but they may improve hemostasis temporarily.
Platelet transfusions may be administered if the presentation is complicated by serious (intracranial) bleeding. Transfused platelets are expected to be cleared very rapidly in the presence of antiplatelet antibodies, but they may improve hemostasis temporarily.
![]() Immunize against encapsulated bacterial organisms (pneumococcus, Haemophilus influenzae, meningococcus) before prolonged immunosuppressive therapy in preparation for splenectomy if required at a later time point.
Immunize against encapsulated bacterial organisms (pneumococcus, Haemophilus influenzae, meningococcus) before prolonged immunosuppressive therapy in preparation for splenectomy if required at a later time point.
![]() Second-Line Treatment (Table 19.3). Despite a high initial response rate (60%–75%), the majority of adults with ITP experience relapse and develop chronic thrombocytopenia once initial treatments are reduced or discontinued. Treatment is appropriate for patients with platelet counts <30,000/μL or clinically significant bleeding.4 The selection of specific therapies should take into account patient preference; some individuals prefer sequential medical therapies prior to undergoing splenectomy, but splenectomy may be preferable in cases of very severe thrombocytopenia associated with bleeding because of a typically rapid postoperative increase in the platelet count in most responding patients.
Second-Line Treatment (Table 19.3). Despite a high initial response rate (60%–75%), the majority of adults with ITP experience relapse and develop chronic thrombocytopenia once initial treatments are reduced or discontinued. Treatment is appropriate for patients with platelet counts <30,000/μL or clinically significant bleeding.4 The selection of specific therapies should take into account patient preference; some individuals prefer sequential medical therapies prior to undergoing splenectomy, but splenectomy may be preferable in cases of very severe thrombocytopenia associated with bleeding because of a typically rapid postoperative increase in the platelet count in most responding patients.
![]() IVIg and anti-D (see above) typically must be readministered every 2 to 3 weeks in most instances.
IVIg and anti-D (see above) typically must be readministered every 2 to 3 weeks in most instances.
![]() The thrombopoietin receptor agonists eltrombopag (starting dose 50 mg orally daily; 25 mg daily in individuals of Asian descent) or romiplostim (starting dose 1 μg/kg SC weekly) produce platelet responses (≥50,000/μL) in approximately 70% of patients with chronic ITP, and they generally are well tolerated.5,6 Potential complications include reticulin deposition in the bone marrow, thrombocytosis, and thrombosis.
The thrombopoietin receptor agonists eltrombopag (starting dose 50 mg orally daily; 25 mg daily in individuals of Asian descent) or romiplostim (starting dose 1 μg/kg SC weekly) produce platelet responses (≥50,000/μL) in approximately 70% of patients with chronic ITP, and they generally are well tolerated.5,6 Potential complications include reticulin deposition in the bone marrow, thrombocytosis, and thrombosis.
![]() The monoclonal anti-B cell antibody rituximab (anti-cd20), given at a dose of 375 mg/m2 weekly for 4 weeks, induces initial and long-term responses in about 50% and 25%, respectively, of adults with severe, chronic ITP. 7
The monoclonal anti-B cell antibody rituximab (anti-cd20), given at a dose of 375 mg/m2 weekly for 4 weeks, induces initial and long-term responses in about 50% and 25%, respectively, of adults with severe, chronic ITP. 7
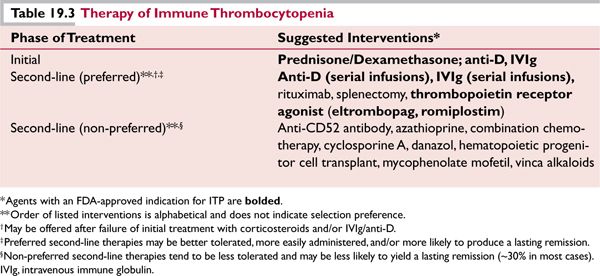
![]() Splenectomy (preferably laparoscopic) yields an immediate response rate of 70% to 75% and durable response rates of 60% to 70%. All patients must receive immunization against encapsulated bacterial organisms (pneumococcus, H. influenzae, meningococcus) several weeks prior to splenectomy if possible. 4
Splenectomy (preferably laparoscopic) yields an immediate response rate of 70% to 75% and durable response rates of 60% to 70%. All patients must receive immunization against encapsulated bacterial organisms (pneumococcus, H. influenzae, meningococcus) several weeks prior to splenectomy if possible. 4
![]() Secondary ITP. A variety of autoimmune, infectious, inflammatory, or malignant conditions may underlie a presentation of ITP.3
Secondary ITP. A variety of autoimmune, infectious, inflammatory, or malignant conditions may underlie a presentation of ITP.3
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


