The development of JAK2 inhibitors followed the discovery of activating mutation of JAK2 (JAK2V617F) in patients with classic Philadelphia-negative myeloproliferative neoplasms (Ph-negative MPN). It is now known that mutations activating the JAK-STAT pathway are ubiquitous in Ph-negative MPN, and that the deregulated JAK-STAT pathway plays a central role in the pathogenesis of these disorders. JAK2 inhibitors thus are effective in patients both with and without the JAK2V617F mutation. This article reviews the rationale for using JAK2 inhibitors in Ph-negative MPN, and the results of more recent clinical trials with these drugs.
- •
Activation of JAK-STAT pathway is at the center of pathogenesis of most cases of Philadelphia-negative myeloproliferative neoplasms.
- •
Activation of JAK-STAT in these disorders is dependent on JAK2, which can be activated either directly or indirectly through activating mutations of JAK2 and related molecules.
- •
Therapy with JAK2 inhibitors in myelofibrosis is associated with significant improvements in splenomegaly, constitutional symptoms, leukocytosis, and thrombocytosis.
- •
No significant improvement in bone marrow fibrosis and JAK2 allelic burden is usually seen with these drugs.
- •
Cytopenias are the most common side effects observed with JAK2 inhibitors.
Introduction
Philadelphia-negative myeloproliferative neoplasms (Ph-negative MPN) are a group of neoplasms that share some common features, including increased tendency to thrombosis and hemorrhage and an increased risk of transforming to acute myeloid leukemia (AML). The classic Ph-negative MPN include polycythemia vera (PV), essential thrombocythemia (ET), and primary myelofibrosis (PMF). MF can also develop secondary to PV (post-PV MF) and ET (post-ET MF).
In 2005, 4 independent groups reported on the presence of an activating mutation of the JAK2 gene (JAK2V617F) in approximately 90% of patients with PV and 60% of patients with ET and MF. The JAK2V617F mutation leads to constitutive activation of the JAK2 tyrosine kinase (TK) and increased signaling through the JAK-STAT (Signal Transducer and Activator of Transcription) pathway, causing hyperresponsiveness to cytokine signaling, increased cellular proliferation, resistance to apoptosis, and DNA damage. Following discovery of the JAK2V617F, other mutations were discovered in patients with JAK2V617F-negative MPN, including JAK2 exon 12 mutations (in 3% of PV patients), MPL W515K/L (in 5%–10% of patients with ET and MF), and CBL mutations (in 6% of MF patients). These mutations share the common theme of leading to deregulation of the JAK-STAT pathway, further underscoring the importance of this pathway in the pathogenesis of Ph-negative MPN.
JAK2V617F formed the rationale for the development of TK inhibitors (TKI) for treating patients with Ph-negative MPN, akin to imatinib and other successfully developed TKI for treatment of malignant neoplasms. The first clinical trials started in mid-2007, and in November 2011 the US Food and Drug Administration (FDA) approved the first JAK2 inhibitor (ruxolitinib; Jakafi) for treatment of MF, which is also the first drug to be approved for this disorder. Although these drugs do not eradicate the neoplastic clone, as is seen with imatinib in chronic myelogenous leukemia, they lead to significant improvements in splenomegaly and systemic symptoms, and may possibly improve survival of patients with MF. Thus, there is great benefit to be gained from these compounds. This article reviews the rationale for using these drugs, and the most recent clinical results.
The JAK kinases
The JAK family of TK includes JAK1, JAK2, JAK3, and TYK2. These kinases were discovered in 1989 and were named after the two-faced Roman god Janus. Structurally, JAK kinases share some common domains ( Fig. 1 ). The JH1 domain is the TK domain, responsible for the phosphorylating activity of JAK kinases. The adjacent JH2 domain, also called the pseudokinase domain, regulates the activity of the JH1 domain. Mutant JAK kinases lacking the JH2 domain display increased TK activity and phosphorylation of downstream messengers such as STAT molecules. It was thought that the JH2 domain did not have true kinase activity. Recent evidence, however, has proved this to be not true. The JH2 domain is a dual-specificity kinase that phosphorylates key residues S523 and Y570, inhibiting the JH1 TK activity. The V617F mutation locates in the JH2 domain and leads to loss of its kinase activity, thus explaining why JAK2V617F causes increased activity of the JAK2 kinase.
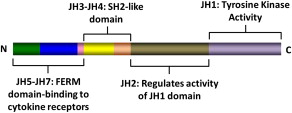
JAK kinases associate with the intracellular portion of cytokine receptors that lack intrinsic TK activity, such as receptors for erythropoietin (EPOR), thrombopoietin (MPL), and interferon (IFNR). Binding of the putative ligand leads to receptor dimerization and activation of associated JAK kinases, followed by activation of multiple intracellular signaling pathways. The various JAK kinases bind to different cytokine receptors. Animal models and reports of patients with germline JAK mutations have proved to be very instructive in our understanding of the specificity of these kinases. JAK1 mutations are essential for signaling through both interferon type-1 (IFN-α/β) and type-2 (IFN-γ) receptors, and also receptors containing glycoprotein 130 (gp130; eg, receptors for interleukin [IL]-6 and related cytokines) and the common γc chain (eg, receptors for IL-2, IL-7, IL-9, and IL-15). JAK2 is essential for erythropoiesis, as mice embryo devoid of JAK2 die during embryonic life owing to failure of developing hematopoiesis. Accordingly, JAK2 is associated with receptors for EPO, thrombopoietin, and receptors using the common β chain (eg, IL-3 receptor, granulocyte-macrophage colony-stimulating factor [GM-CSF] receptor), which are essential for granulopoiesis. The JAK3 TK is associated with receptors of the IL-2 receptor family that share the common γc chain. JAK3 deficiency impairs development of both T and B lymphocytes, as well as natural killer (NK) cells, and JAK3 mutations have been described in patients with severe combined immunodeficiency, a form of primary immunodeficiency. TYK2 mediates signaling of receptors for type-1 IFN (IFN-α/β) and IL-12.
The JAK kinases
The JAK family of TK includes JAK1, JAK2, JAK3, and TYK2. These kinases were discovered in 1989 and were named after the two-faced Roman god Janus. Structurally, JAK kinases share some common domains ( Fig. 1 ). The JH1 domain is the TK domain, responsible for the phosphorylating activity of JAK kinases. The adjacent JH2 domain, also called the pseudokinase domain, regulates the activity of the JH1 domain. Mutant JAK kinases lacking the JH2 domain display increased TK activity and phosphorylation of downstream messengers such as STAT molecules. It was thought that the JH2 domain did not have true kinase activity. Recent evidence, however, has proved this to be not true. The JH2 domain is a dual-specificity kinase that phosphorylates key residues S523 and Y570, inhibiting the JH1 TK activity. The V617F mutation locates in the JH2 domain and leads to loss of its kinase activity, thus explaining why JAK2V617F causes increased activity of the JAK2 kinase.
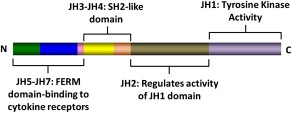
JAK kinases associate with the intracellular portion of cytokine receptors that lack intrinsic TK activity, such as receptors for erythropoietin (EPOR), thrombopoietin (MPL), and interferon (IFNR). Binding of the putative ligand leads to receptor dimerization and activation of associated JAK kinases, followed by activation of multiple intracellular signaling pathways. The various JAK kinases bind to different cytokine receptors. Animal models and reports of patients with germline JAK mutations have proved to be very instructive in our understanding of the specificity of these kinases. JAK1 mutations are essential for signaling through both interferon type-1 (IFN-α/β) and type-2 (IFN-γ) receptors, and also receptors containing glycoprotein 130 (gp130; eg, receptors for interleukin [IL]-6 and related cytokines) and the common γc chain (eg, receptors for IL-2, IL-7, IL-9, and IL-15). JAK2 is essential for erythropoiesis, as mice embryo devoid of JAK2 die during embryonic life owing to failure of developing hematopoiesis. Accordingly, JAK2 is associated with receptors for EPO, thrombopoietin, and receptors using the common β chain (eg, IL-3 receptor, granulocyte-macrophage colony-stimulating factor [GM-CSF] receptor), which are essential for granulopoiesis. The JAK3 TK is associated with receptors of the IL-2 receptor family that share the common γc chain. JAK3 deficiency impairs development of both T and B lymphocytes, as well as natural killer (NK) cells, and JAK3 mutations have been described in patients with severe combined immunodeficiency, a form of primary immunodeficiency. TYK2 mediates signaling of receptors for type-1 IFN (IFN-α/β) and IL-12.
JAK kinases as central molecules in the pathogenesis of Ph-negative MPN
The first hints for a role of JAK kinases in MPN came from studies conducted in the Drosophila melanogaster fruit fly. It was found that an activating mutation (E695K) in the JH2 domain of the protein encoded by the Hopscotch gene, the JAK equivalent in Drosophila , led to an increased proliferation of hemocytes (fly blood cells) and a clinical picture reminiscent of a leukemia. Increased kinase activation was demonstrated, as well as increased phosphorylation of downstream target STAT92E.
Experimental studies demonstrated that the JAK2V617F oncogenic mutation leads to increased cellular proliferation and resistance to apoptosis. Expression of JAK2V617F in Ba/F3 cells expressing EPOR leads to increased cell proliferation and hyperresponsiveness to EPO. Several animal models of JAK2V617F-positive MPN have been published. Mice harboring hematopoietic stem cells and progenitor cells expressing JAK2V617F develop a PV-like disease with bone marrow hypercellularity, increased hematocrit, and splenomegaly, and some mice eventually develop a clinical picture compatible with MF.
The phenotype acquired with the JAK2V617F mutation is secondary to activation of intracellular oncogenic signaling pathways. Central among these is the JAK-STAT pathway. JAK2V617F phosphorylates latent cytoplasmic transcription factors STAT3 and STAT5. This process leads to STAT dimerization and translocation to the nucleus where they induce expression of several genes relevant to the neoplastic phenotype, including CCND1 , BCLXL , and BIRC5A . The role of STATs as central mediators of JAK2-aberrant signaling in MPN has been demonstrated in recent publications. In a PV mouse model, inducible deletion of STAT5 through a Cre-recombinase–mediated mechanism essentially abolished all signs and symptoms of the disease, with normalization of the red cell mass, splenomegaly, and bone marrow cellularity. Rescuing the STAT5 deletion with expression of only one allele restored disease features, underscoring the important role of STAT5 in the pathophysiology of MPN. Other reports have confirmed that STAT5 is an essential mediator of JAK2V617F-mutated disease. Besides STATs, JAK2V617F can activate the oncogenic pathways PI3K-Akt-mTOR (phosphatidylinositol 3-kinase–Akt–mammalian target of rapamycin) and Ras-Raf-MEK-ERK (MAPK; mitogen-activated protein kinases). Patients with JAK2V617F-negative MPN also have increased STAT5, Akt, and Erk phosphorylation, demonstrating that activation of these pathways is a common feature of all patients with Ph-negative MPN.
JAK kinases may also contribute to oncogenesis by inducing epigenetic changes in the genome. It has been demonstrated that activated JAK can be found in the nucleus of hematopoietic cells, where it phosphorylates histone H3 at residue Y41. This process inhibits binding of heterochromatin protein 1α (HP1α), which is responsible for gene silencing through epigenetic mechanisms. This epigenetic deregulation induced by JAK2 increases expression of oncogene LMO2 , and inhibiting JAK2 increased HP1α phosphorylation and decreased LMO2 expression. Nuclear JAK2 has been demonstrated in the CD34 + cells of patients with Ph-negative MPN. Thus, JAK2 may regulate gene expression not only through activation of oncogenic molecules, such as STAT5, but also through epigenetic deregulation.
More recently, the role of cytokines has gained greater importance in the pathophysiology of Ph-negative MPN, particularly MF. Several proinflammatory and profibrotic cytokines (eg, transforming growth factor β, IL-1b, IL-2, IL-6, IL-8, IL-12, IL-15, tumor necrosis factor α [TNF-α]) have been found to be elevated in patients with MF and PV. Cells that are responsible for cytokine production include neoplastic megakaryocytes, monocytes, and bone marrow stromal cells. These cytokines are associated with many of the clinical features of Ph-negative MPN, including bone marrow fibrosis, osteosclerosis, constitutional symptoms, hematopoietic stem cell mobilization, and transfusion-dependent anemia. In one recent report, increased levels of cytokines IL-8, IL-2 receptor, IL-12, IL-15, and IP-10 (IFN-γ–inducible protein 10) were found to be associated with decreased overall survival in patients with MF. Several of these cytokines are dependent on the JAK-STAT for intracellular signaling, and STAT3 activation increases autocrine production of proinflammatory cytokines such as IL-6. In addition, increased cytokine signaling may lead to resistance to JAK2 inhibitors. Knock-down of the JAK2V617F gene with small interfering RNA inhibited proliferation of JAK2V617F-positive cells or CD34 + cells from patients with MPN. However, addition of IL-3 and thrombopoietin impeded growth inhibition and increased STAT5 activation. In another study, coculture of JAK2V617F cells with bone marrow stromal cells blocked JAK2 inhibition by the compound atiprimod. This protective effect of stromal cells was due to their production of proinflammatory cytokines IL-6 and IP-10.
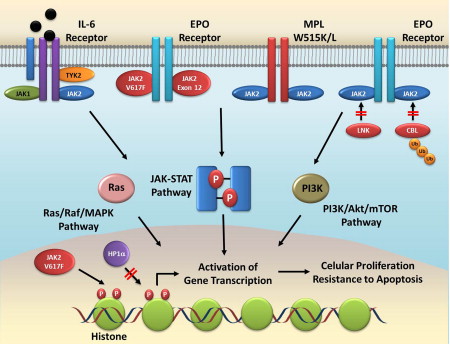
In conclusion, the following picture emerges from our current understanding of the pathophysiology of Ph-negative MPN ( Fig. 2 ). These disorders are caused by mutations that lead to chronic, persistent activation of the JAK-STAT pathway in hematopoietic stem cells. Mutations can either directly activate the JAK2 kinase (eg, JAKV617F, JAK2 exon 12 mutation) or indirectly (eg, MPL mutation, CBL mutation). Activation of the JAK-STAT pathway leads to increased cellular proliferation, resistance to apoptosis, genetic instability, and acquisition of further mutations. Epigenetic effects of JAK activation and the balance between STAT1 and STAT5 activation are likely related to the different disease phenotypes associated with these various mutations. Chronic JAK-STAT activation also leads to increased production of proinflammatory cytokines, which further contribute to disease pathogenesis and activation of the pathway. Although the JAK2V617F mutation is not detected in all patients with Ph-negative MPN, activation of JAK kinases (mutated or wild-type) remains at the center of the pathogenesis of probably most patients with these disorders. Thus, the development of drugs with ability to inhibit chronic JAK-STAT signaling is an important step toward achieving effective therapeutic agents for these patients.
Results of JAK2 inhibitors in myelofibrosis
Several JAK2 inhibitors are currently under development for the treatment of Ph-negative MPN ( Table 1 ). These inhibitors differ in their specificity against JAK2 kinases, and their main therapeutic benefits in patients with MF are reduction of splenomegaly and improvement in systemic symptoms.
| Compound | Disease | Target | Responses | Toxicity |
|---|---|---|---|---|
| Ruxolitinib | MF | JAK1, JAK2 | Splenomegaly 28%–44% Improvement in MF-related symptoms, leukocytosis, thrombocytosis, overall survival | Anemia, thrombocytopenia, dizziness Headache |
| Ruxolitinib | PV/ET | JAK1, JAK2 | PV-ORR 97% (CR 50%) ET-ORR 90% (CR 26%) | Anemia, thrombocytopenia, dizziness Headache |
| SAR302503 | MF | JAK2 | Splenomegaly 39% Improvement in MF-related symptoms, leukocytosis, thrombocytosis, JAK2V617F allelic burden | Anemia, thrombocytopenia, diarrhea Nausea, vomiting, elevated pancreatic enzymes |
| CYT387 | MF | JAK1, JAK2, TYK2 | Splenomegaly 31% Improvement in MF-related symptoms Transfusion independence 46% | Thrombocytopenia, first-dose effect, peripheral neuropathy |
| Pacritinib | MF | JAK2, FLT3 | Splenomegaly 39% Improvement in MF-related symptoms | Diarrhea, nausea, vomiting Can be safely used in patients with cytopenias |
| LY2784544 | MF | JAK2V617F | Splenomegaly 41% Improvement in MF-related symptoms, bone marrow fibrosis | Tumor lysis syndrome, creatinine increase |
| NS-018 | MF | JAK2, SRC kinases | NA | NA |
| BMS-911453 | MF | JAK2 | NA | NA |
Ruxolitinib
Ruxolitinib (formerly known as INCB018424; Incyte, Wilmington, DE) is an orally available, dual JAK1 and JAK2 inhibitor that has been recently approved as therapy for patients with intermediate or high-risk MF, either primary or post-PV/ET. In preclinical evaluation, ruxolitinib inhibited both JAK1 (half-maximal concentration [IC 50 ] = 3.3 nM) and JAK2 (IC 50 = 2.8 nM), while sparing JAK3 (IC 50 = 322 nM). Ruxolitinib inhibited proliferation of JAK2V617F-positive cell lines and CD34 + cells from patients with MPN, and this was associated with decreased phosphorylation of STAT3. In mouse models, ruxolitinib led to spleen reduction, weight gain, and improved cytokine profile.
A phase I/II clinical trial recruited 153 patients with MF to be treated with ruxolitinib at different doses and schedules. The dose-limiting toxicity (DLT) was thrombocytopenia, and maximum tolerated doses were 25 mg twice daily or 50 mg once daily. However, a dose-adjusted schedule whereby patients started with 15 mg twice daily (10 mg if platelet count between 100 × 10 9 /L and 200 × 10 9 /L) with further monthly dose increments if there was no response or side effect led to the best balance between efficacy and toxicity. A total of 61 among 140 patients had a clinical improvement in spleen size by the International Working Group for Myelofibrosis Research and Treatment (IWG-MRT) response criteria (ie, ≥50% spleen size reduction by physical examination). This figure corresponded to a 35% decrease in spleen volume by magnetic resonance imaging (MRI). Responses were observed in JAK2V617F-positive and -negative patients. After 1 year, 73% of patients who started with 15 mg twice daily and 78% of patients who started with 25 mg twice daily maintained their response. There was substantial improvement in systemic symptoms, including night sweats, pruritus, fatigue, bone pain, and abdominal pain, as well as improved ability to walk. Improvement in symptoms was accompanied by a decrease in proinflammatory cytokine levels (eg, TNF-α, IL-6, IL-1ra). Reduction in intracellular phosphorylated STAT3 was observed after treatment with ruxolitinib, consistent with on-target activity.
More recently, 2 reports have focused on longer follow-up of patients who were enrolled in the phase I/II trial. In one report, the outcomes of 107 patients with MF who were enrolled and treated at M.D. Anderson Cancer Center (MDACC) was compared with an historical cohort of 310 patients identified from 3 large MPN databases. The historical cohort was matched to the MDACC cohort based on eligibility criteria for the phase I/II trial. In the MDACC cohort, after a median follow-up of 32 months, 54% of patients remained on study. Median duration of spleen response had not been reached at the time of report. Most common reasons for discontinuation were death (12.1%) and progressive disease (11.2%). Compared with the historical cohort, therapy with ruxolitinib significantly improved survival outcomes, with a hazard ratio (HR) of 0.61 (95% confidence interval [CI] 0.41–0.89; P = .022) for overall survival (OS). This effect was more pronounced in the subgroup of patients at high risk according to the International Prognostic Scoring System (IPSS) (HR = 0.50; 95% CI 0.31–0.81; P = .006). By contrast, the report from the Mayo Clinic on 51 patients treated with ruxolitinib at that center did not reveal an improvement in OS compared with a cohort of 410 patients with MF from all IPSS risk categories treated with standard therapy (unadjusted P = .43; P = .58 after adjusting for the Dynamic IPSS-Plus score). The rate of treatment discontinuation was very high in the Mayo Clinic cohort (51% at 1 year, 89% at 3 years), and this may have affected the results obtained. Most common reasons for discontinuation in the Mayo cohort were patient withdrawal of consent (29.4%), physician decision (23.5%), and disease progression (19.6%).
Ruxolitinib was further evaluated in 2 phase III trials for patients with MF. In the trial conducted in North America and Australia (COMFORT-I), 309 patients with MF and Int-2/High IPSS risk were randomized among ruxolitinib and placebo in a 1:1 fashion. Starting dose was 15 mg twice daily for those patients with a platelet count between 100 and 200 × 10 9 /L, and 20 mg twice daily for those patients with a platelet count higher than 200 × 10 9 /L. The primary end point was the percentage of patients with a reduction in spleen volume by MRI of at least 35% at 24 weeks. After a median follow-up of 32 weeks, the primary end point had been reached in 41.9% of patients in the ruxolitinib group versus 0.7% of patients in the placebo group ( P <.0001). Patients receiving ruxolitinib had a mean decrease in spleen volume of 31.6% versus an increase of 8.1% in patients receiving placebo. Similar to phase I/II trial data, there were significant improvements in symptoms related to MF. The Myelofibrosis Symptom Assessment Form Total Symptom Score (TSS; sum of scores for itching, night sweats, bone/muscle pain, abdominal discomfort, pain under the ribs on the left, and early satiety) decreased by 50% or greater in 46% of patients in the ruxolitinib arm versus 5.3% of patients in the placebo arm ( P <.001). The efficacy of ruxolitinib in decreasing splenomegaly and improving systemic symptoms was independent of the presence of the JAK2V617F mutation. There was reduction in proinflammatory cytokines, and an increase in leptin and EPO levels. After a median follow-up of 51 weeks, a planned data cutoff analysis for safety revealed that ruxolitinib improved survival compared with placebo (HR 0.5; P = .04).
In the companion trial COMFORT-II, 219 patients with Int-2/High-risk MF were randomized 2:1 to receive ruxolitinib or best available therapy. The primary end point was percentage of patients with a reduction of at least 35% in spleen volume by MRI at 48 weeks. In the ruxolitinib arm, 28% of patients reached the primary end point, versus 0% in control arm ( P <.001). Responses were durable, and after 12 months of follow-up 80% of responding patients receiving ruxolitinib still maintained a response. There was also significant improvement in quality of life and symptoms associated with MF, including fatigue, insomnia, and appetite loss in patients receiving ruxolitinib, whereas a worsening of these symptoms were observed in patients in the best available therapy group. There was no improvement in OS in patients receiving ruxolitinib, but the study was underpowered to detect such a difference.
Ruxolitinib is a relatively well tolerated medication. Most nonhematologic side effects are grade 1 to 2 in severity, and treatment discontinuations attributable to side effects were uncommon in both phase III trials (11% and 8%). Side effects that occurred more frequently with ruxolitinib than with placebo in the COMFORT-I study included ecchymosis (18.7% vs 9.3%), headache (14.8% vs 5.3%), and dizziness (14.8% vs 6.6%). These side effects were all grade 1 to 2 in severity, with the exception of one case of dizziness grade 3 to 4. Because of its mechanism of action, cytopenias are a common side effect of ruxolitinib. Compared with placebo, anemia grade 3 to 4 occurred in 25% more patients in the ruxolitinib arm of the COMFORT-I phase III trial, while thrombocytopenia grade 3 to 4 was documented in an excess of 12% of cases. Severe (grade 3–4) neutropenia was uncommon (7% in one trial). Data from both phase III trials demonstrated a clear time-dependent pattern of anemia severity during therapy with ruxolitinib: hemoglobin drops approximately 1.5 g/dL during the first 8 to 12 weeks of therapy, reaching a mean nadir value of 9.4 g/dL. By 24 weeks of therapy, hemoglobin increased to a new mean steady state of 10.1 g/dL. This pattern was independent of transfusions and dose modifications. Most cases of thrombocytopenia grade 3 to 4 happened during the first 8 weeks of therapy with ruxolitinib, and were managed with dose reductions and treatment interruptions. The monthly prevalence of thrombocytopenia decreased to placebo levels with 24 weeks of therapy. Bleeding episodes of grade 3 to 4 happened in 3.9% of patients being treated with ruxolitinib and 3.3% of patients in the placebo arm. Despite its high incidence, few patients (only 1 in each trial) discontinued ruxolitinib because of cytopenias.
Patients who discontinue ruxolitinib have a rapid relapse of symptoms, usually within 7 days of therapy interruption. One report has stated that patients who discontinue therapy with ruxolitinib may develop a cytokine rebound syndrome, with acute spleen enlargement and a shock-like syndrome. In the COMFORT-I trial, adverse events after treatment interruption were not more commonly seen with ruxolitinib. Grade-3 events occurred in 16% of patients in the ruxolitinib arm compared with 13% of patients in the placebo arm. No clear pattern suggestive of a withdrawal effect was seen. Severe adverse events occurred in only 3 patients in each treatment arm (6.1% [ruxolitinib] vs 5.6% [placebo]). Eleven percent of patients in each arm of the COMFORT-I study discontinued therapy because of side effects.
The FDA approved ruxolitinib for the treatment of patients with intermediate-risk and high-risk MF. Despite this, ruxolitinib is still being evaluated in clinical trials. Alternative dose schedules for patients with thrombocytopenia (50–100 × 10 9 /L) are being studied ( NCT01348490 , NCT01317875 ), as well as a sustained-release formulation ( NCT01340651 ). Combination trials with lenalidomide ( NCT01375140 ) and panobinostat ( NCT01433445 ) have recently started.
SAR302503
SAR302503 (formerly known as TG101348; Sanofi S.A., Paris, France) is an orally available TKI that has selective activity versus JAK2 (IC 50 = 3 nM). SAR302503 has limited activity against JAK1 (IC 50 = 105 nM) and no activity against JAK3 (IC 50 = 996 nM). Preclinical evaluation of SAR302503 demonstrated it to be a potent inhibitor of JAK2V617F oncogenic signaling, and it had in vivo activity in a mouse model of JAK2V617F-positive MPN.
SAR302503 has been evaluated in a phase I/II clinical trial that recruited 59 patients with MF, the majority of whom (86%) was JAK2V617F-positive. The maximum tolerated dose (MTD) was determined to be 680 mg once daily, and DLT included asymptomatic hyperamylasemia and hyperlipasemia grade 3 to 4. After 6 cycles of therapy, a spleen response by IWG-MRT criteria was seen in 39% of patients, and the spleen response was 45% in the MTD cohort. Median time to response was 113 days and the mean duration of spleen response was 315 days. SAR302503 led to normalization of leukocytosis and thrombocytosis in 56% and 90% of cases, respectively. The drug led to improvements in systemic symptoms, including cough, early satiety, fatigue, night sweats, and pruritus. No decrease in proinflammatory cytokines was observed in this trial, suggesting that this drug has more of an antiproliferative than an anticytokine effect. A decrease in JAK2V617F allele burden has been reported with this drug. At baseline, the median allele burden was 20% (range 3%–100%), which decreased to 9% (range 0%–100%; P = .03) after 24 cycles of treatment. A similar effect was seen in patients who presented with baseline JAK2V617F burden greater than 20% (baseline median 60% [range 23%–100%]; after 24 cycles of therapy, median 21% [range 6%–100%]; P = .03).
Toxicities of SAR302503 include cytopenias, gastrointestinal toxicity, and elevation of pancreatic enzymes. In the phase I/II study, anemia was reported in 43.2% of patients, and it was grade 3 to 4 in 35% of cases. Thrombocytopenia occurred in 40.7% (grade 3–4 23.7%) and neutropenia in 4.4% (grade 3–4 1%). Among nonhematological adverse events, diarrhea (all grades 64%, grade 3–4 10%), nausea (all grades 69.5%, grade 3–4 3.4%), and vomiting (all grades 57.6%, grade 3–4 3.4%) were the most commonly reported. In the MTD cohort of patients (n = 40), 70% of patients required dose reductions during the first 6 cycles of therapy, more commonly because of gastrointestinal toxicity or cytopenias (32.5% of cases).
SAR302503 is highly effective for treating patients with MF, but has a relatively high incidence of side effects at the MTD. A lower starting dose may provide similar efficacy with more manageable toxicity. At present, a randomized trial ( NCT01437787 ) of SAR302503 versus placebo for patients with Int-2/high-risk MF is recruiting, and patients randomized to the drug will receive SAR302503 at doses of 400 or 500 mg once daily.
CYT387
The compound CYT387 (YM Biosciences, Mississauga, Canada) is an orally available JAK1 (IC 50 = 11 nM), JAK2 (IC 50 = 17 nM), and TYK2 (IC 50 = 17 nM) inhibitor. Preclinical activity of CYT387 was demonstrated against both JAK2V617F- and MPL-mutated cell lines.
A phase I/II clinical trial of CYT387 reported on 166 patients with MF (primary = 65%; post-PV = 22%; post-ET = 14%) treated with CYT387 at different doses and schedules. The MTD was 300 mg once daily, and DLTs were headache and hyperlipasemia. After a median follow-up of 10.4 months, the response rate on splenomegaly by IWG-MRT criteria was 31%. Responses were rapid, occurring at a median of 15 days after the start of treatment. There was also significant improvement in MF-related symptoms, including bone pain (49% improvement), cough (49%), fever (100%), night sweats (80%), and pruritus (74%). An improvement in erythropoiesis and transfusion dependency has been seen with this drug. Among 68 patients (41%) who were transfusion-dependent at baseline, according to IWG-MRT criteria, 46% achieved transfusion independence and a hemoglobin level of 8 g/dL or higher. Median time to transfusion independence was 84 days. Median duration has not been reached.
Similar to other compounds of this class, CYT387 can induce thrombocytopenia (all grades 33%, grade 3–4 17%). Neutropenia and anemia have also been reported, albeit at much lower rates (grade 3–4 3% and 1%, respectively). The first dose effect is a transient dizziness and lightheadedness that may be accompanied by hypotension, and occurs after the patient takes the first dose of the drug. It has been reported in 20% of patients, and is usually grade 1 in severity. Peripheral neuropathy (grade 1–2 only, 20%), nausea (grade 1–2 17%), and diarrhea (grade 1–2 18%) have also occurred with this medication.
The compound CYT387 is highly effective for inducing spleen responses and improvement in constitutional symptoms, while possessing a relatively favorable side-effect profile, similar to other JAK2 inhibitors. The reported rates of transfusion independence are encouraging and need to be further explored in additional trials. Despite this, it should be mentioned that the IWG-MRT criteria for transfusion independence are suboptimal, because in the COMFORT-I trial a transfusion independence rate of 47% was reported in patients in the placebo, underscoring the need for better and improved criteria.
Other JAK2 Inhibitors in Clinical Development
The JAK2 inhibitor pacritinib (formerly known as SB1518; Cell Therapeutics, Inc, Seattle, WA) has selective activity against JAK2 (IC 50 = 19 nM) and is in clinical trials at the moment. Phase I studies determined the MTD to be 500 mg/d, but pharmacokinetic analysis recommended a dose of 400 mg/d because no increase in plasma level is seen above this dosage. A phase II study has recruited 31 patients, and recently presented data demonstrated that pacritinib improves splenomegaly (39% by physical examination; 55% had a ≥25% reduction in spleen volume by MRI) and constitutional symptoms (31%–78% improvement in Myelofibrosis Symptom Assessment Form scores by the 7th–10th cycle). Adverse events consisted mostly of diarrhea (all grades 87%; grade 3–4 10%), nausea (all grades 45%; grade 3–4 6%), and vomiting (all grades 29%; grade 3–4 3%). Pacritinib appears to induce few treatment-related cytopenias. Rates of thrombocytopenia and anemia grade 3 to 4 are low (14% and 5%, respectively), and the drug could be safely used in patients with a platelet count less than 100 × 10 9 /L. Spleen response was not affected by pretreatment platelet counts (35% [platelet <150 × 10 9 /L] vs 39% [all patients]).
LY2784544 (Eli Lilly, Indianapolis, IN) is an orally available, selective inhibitor of JAK2V617F (IC 50 = 55 nM), harboring no activity against wild-type JAK2 (IC 50 = 2.26 μM). Preclinical studies demonstrated that LY2784544 inhibited JAK2V617F and STAT5 phosphorylation, induced cell-cycle arrest and apoptosis, and reduced growth of Ba/F3-JAKV617F-GFP tumor cells xenografts in mice. At the same time, LY2784544 did not inhibit proliferation of cells expressing wild-type JAK2 (IC 50 = 1.356 μM). Preliminary results of a phase I clinical trial of LY2784544 have been presented in abstract form. Nineteen patients (MF = 18, PV = 1) were recruited, and the MTD was reported to be 120 mg. DLTs included hyperuricemia and creatinine increase. There were 4 cases of tumor lysis syndrome, 3 of which had clinical consequences. Regarding response rates, of 17 evaluable patients, 41% had a reduction in splenomegaly of 50% or greater. A reduction in MPN-related symptoms of 50% or more was observed in 59% of patients. In addition, very preliminary observations suggest that an improvement in bone marrow fibrosis might be obtained, as 3 of 5 patients with follow-up bone marrow biopsies had a reduction in the severity of marrow fibrosis. The study is ongoing and alternative dose schedules to decrease toxicity will be explored.
NS-018 (Nippon Shinyaku Co. Ltd, Kyoto, Japan) is a JAK2-specific inhibitor that also has activity against kinases of the Src family. In vitro, NS-018 inhibits JAK2 (IC 50 = 0.72 nM), has limited activity against JAK1, JA3, and TYK2, and can inhibit several Src-family kinases including SRC, FYN, and YES. NS-018 inhibits JAK2 phosphorylation in Ba/F3 cells expressing EPOR and JAK2V617F, and demonstrated activity in a mouse model of JAK2V617F-induced MPN, with resolution of splenomegaly, extramedullary hematopoiesis, and leukocytosis. NS-018 is being tested in a phase I/II clinical trial in patients with MF (NCT01423851).
BMS-911543 (Bristol-Myers Squibb, New York, NY) is a JAK2 kinase selective inhibitor (IC 50 = 1 nM) that has virtually no activity against other JAK family members and other target kinases. BMS-911543 inhibited proliferation of cells expressing JAK2V617F, and had no effect in cell lines dependent on JAK3 for proliferation. The drug inhibited proliferation of primary CD34 + cells from patients with MPN (IC 50 = 0.15–0.9 μM) while having a limited effect against control CD34 + cells (IC 50 = 1.5 μM). A phase I/II clinical trial in patients with MF (NCT01236352) will provide further information on the clinical activity of BMS-911543.
Related posts:
 Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Role of Germline Genetic Factors in MPN Pathogenesis
Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Genotype-Phenotype Interactions in the Myeloproliferative Neoplasms
Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Role of Germline Genetic Factors in MPN Pathogenesis
Role of TET2and ASXL1Mutations in the Pathogenesis of Myeloproliferative Neoplasms
Myeloproliferative Neoplasm Animal Models
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


