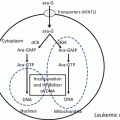
Fig. 20.1
Various causes of LSC development
In recent years, new techniques have begun to revolutionize the diagnosis, prognosis, and classification of leukemia. More recently, the introduction of high-speed analysis of whole-genome sequencing has made it possible to find multiple gene mutations that are involved in leukemogenesis. During the process by which genetic variation accumulates in HSCs, the fraction termed pre-LSCs gradually expands, and then LSCs are fully established when additional gene mutations are acquired. This final trigger is considered to occur in LSCs in some cases and in progenitor cells differentiated from LSCs in other cases.
As mentioned above, AML-LSCs are present in the CD34+CD38− fraction, which is a phenotype shared with normal HSCs. Thus, leukemia originates from normal HSCs. HSCs have a long lifetime and can undergo a considerable number of divisions; therefore, genetic and epigenetic alterations tend to accumulate in these cells. In addition, because HSCs have molecular mechanisms for self-replication, they fulfill the requirements of LSCs when genes that contribute to a growth advantage and a differentiation block are obtained. In principle, differentiated progenitor cells have no self-renewal ability. However, there is a possibility that LSCs originate from these differentiated progenies after they acquire self-renewal ability. To become LSCs, differentiated leukemic progenitors must have self-renewal ability.
The mixed lineage leukemia (MLL) gene present in chromosome 11q23 leads to the development of a variety of diseases by forming various fusion genes such as the MLL/GAS fusion gene formed by the 11;17 chromosomal translocation, the MLL/ENL rearranged gene formed by the 11;19 translocation, and the MLL/AF9 formed by the 9;11 translocation [12, 13, 14]. A study examined whether these fusion genes can lead to the development of leukemia upon their introduction into murine HSCs or progenitor cells. The MLL/GAS7 fusion gene causes leukemia only when introduced into HSCs, while MLL/ENL and MLL/AF9 cause leukemia when introduced not only into HSCs with self-renewal ability but also into GMPs. Similarly, leukemia can develop when the MOZ/TIF2 fusion gene, which is formed by the inversion of chromosome 8, is introduced into GMPs [15]. These findings indicate that it is possible to develop LSCs from precursor cells, although it is dependent on the type of genetic abnormality.
20.4 Candidate Therapeutic Molecules for Targeting LSCs
20.4.1 Signal Transduction and Metabolic Pathways
The therapeutic strategies for eradicating LSCs are summarized in Table 20.1. Molecules that control self-renewal and proliferation, molecules that regulate the cell cycle and differentiation, molecules that enhance cell survival, molecules constituting the hematopoietic (leukemic) niche that exists in hypoxic environments, and cell surface molecules specific for leukemia are candidates for eradicating LSCs. However, many of these molecules are present even in normal HSCs. To specifically eradicate LSCs while sparing normal HSCs, the specific molecular mechanisms in LSCs need to be clarified.
Table 20.1
Therapeutic strategies targeting LSCs
1. Antibodies specific for LSCs |
2. Inhibitors of signal transduction and metabolic pathways |
3. Induction of apoptosis |
4. Suppression of the self-renewal capacity of LSCs |
5. Transition into the cell-cycling state from the quiescent state and differentiation induction |
6. Detachment of LSCs from the hypoxic niche |
7. Targeting the hematopoietic niche or microenvironment |
The canonical Wnt/β-catenin signal is constitutively active in both AML and chronic myeloid leukemia (CML), in addition to many human cancers [16]. Analysis of patient samples demonstrated that LSCs in the chronic phase of CML develop from HSCs, while those in the accelerated and blastic phases of CML develop by transformation of CMPs or GMPs through activation of the Wnt/β-catenin pathway. β- and γ-catenin are not essential at least for the maintenance of murine normal HSCs, and they are therefore potential therapeutic targets in LSCs [17].
The hedgehog (Hh) pathway is abnormally activated in various types of cancer [18]. During embryonic pattern formation, the Hh signaling pathway regulates proliferation and differentiation. In cancer, the pathway increases tumor invasiveness and regular CSC proliferation. In some preclinical models, aberrant activation of the Hh pathway is necessary for the maintenance of some CD34+ LSCs. PF-04449913 is a novel oral small molecule inhibitor that binds to smoothened (SMO), a membrane protein in the Hh pathway [19]. In AML cell lines and human primary AML cells, PF-04449913 appears to improve sensitivity to cytarabine in dormant LSCs. Multiple phase 1 studies are ongoing with PF-04449913 as a monotherapy or in combination with low-dose cytarabine or hypomethylating agents to treat AML [20]. Vismodegib, a SMO antagonist approved for the treatment of basal-cell carcinoma, in combination with ribavirin with or without decitabine, is also under investigation in relapsed/refractory AML (NCT02073838) [20].
20.4.2 Cell Surface Antigens
Identifying cell surface antigens that are differentially upregulated on LSCs but not on normal HSCs has been a major focus. However, it is difficult to identify such antigens, and thus far no unique antigen has been found that is specifically expressed on CD34+CD38− LSCs but not on normal HSCs. Despite these difficulties, many cell surface antigens have been identified that are preferentially expressed on CD34+CD38− LSCs compared with normal human HSCs. These include CD25 [21], CD32 [21], CD33 [22], CD44 [23], CD47 [24, 25], CD96 [26], CD123 [27–29], TIM-3 [30–32], interleukin (IL)-1 receptor accessory protein (IL1RAP) [37], and C-type lectin-like molecule-1 (CLL-1) [38)], as summarized in Table 20.2.
Table 20.2
Possible target candidates of surface antigens expressed on LSCs
Surface antigen | Characteristics | Expressed on | References | |
|---|---|---|---|---|
Normal HSCs | LSCs | |||
CD25 | IL-2Rα chain, expressed on activated T cells | ± | + | [21] |
CD32 | FcγRIIA, expressed on monocytes and neutrophils | ± | + | [21] |
CD33 | Pan-myeloid antigen, clinically used as the antibody gemtuzumab ozogamicin | ++ | ++ | [22] |
CD44 | Adhesion molecule, hyaluronic acid receptor | + | ++ | [23] |
CD47 | The ligand for signal regulatory protein alpha (SIRPα), anti-CD47 antibodies induce apoptosis of AML-LSCs | + | ++ | |
CD96 | Member of the Ig gene superfamily, expressed in T and NK cells, but not in B cells, granulocytes, monocytes, or red blood cells | + | ++ | [26] |
CD123 | IL-3R α chain, expressed on total and CD34+CD38− fractions of AML and normal myeloid cells | + | ++ | |
TIM-3 | T-cell immunoglobulin mucin-3, expressed in CD4+ Th1 lymphocytes and is an important regulator of Th1 cell immunity and tolerance induction | + | ++ | |
IL1RAP | IL1 receptor accessory protein (IL1RAP, IL1R3), expressed on candidate LSCs in the majority of AML patients but not on normal HSCs | −〜 ± ? | + | [37] |
CLL-1 | C-type lectin-like molecule-1, expressed on monocytes, neutrophils, and the majority of AML blasts | ± | + | [38] |
In normal human BM, CD25 is expressed in a portion of CD4+ T cells (regulatory T cells), and CD32 (Fc gamma receptor II) is expressed in B cells, T cells, and monocytes; however, neither antigen is expressed in CD34+CD38−CD133+ HSCs. By demonstrating the functional significance of distinctly expressed genes using analyses including xenotransplantation, it was reported that CD25 and CD32 were most frequently expressed on LSCs of AML patients, 85% of whom were poor risk, and it was suggested that therapeutic strategies targeting these molecules may be effective and improve patient prognosis [21].
CD44 is a cell adhesion molecule whose ligand is hyaluronic acid and which contributes to construction of the LSC niche. The engraftment of human AML into immunodeficient mice is completely inhibited by administration of an anti-CD44 antibody (H90), while that of normal HSCs is only minimally affected [23]. In this paper, Jin et al. reported that CD44 is a key regulator of AML-LSC function and is required for proper homing of AML-LSCs to microenvironments and to maintain AML-LSCs in a leukemic niche. The finding that AML-LSCs require interactions with a niche to maintain their stem cell properties provides a therapeutic strategy to eliminate dormant AML-LSCs and may be applicable to other types of CSCs.
CD47 belongs to the immunoglobulin (Ig) superfamily and is a ligand of SIRPα (signal regulatory protein alpha) expressed on macrophages. When the IgV region of SIRPα expressed on macrophages combines with CD47 on LSCs, a tyrosine residue of SIRPα is phosphorylated, and the inhibitory signals of phagocytosis (“don’t eat me” signal) are transmitted, resulting in the inhibition of phagocytosis by macrophages. On the other hand, when the target cell does not express CD47 or the binding of SIRPα and CD47 is inhibited, the signal of the inhibition is not transmitted, and consequently the target cells are phagocytosed by macrophages. When an anti-CD47 antibody blocks the binding of SIRPα and CD47 expressed on LSCs, the “don’t eat me” signal to macrophages is blocked, and LSCs are phagocytosed. Increased phagocytic action by macrophages is a mechanism of action of anti-CD47 antibodies. CD47 is strongly expressed on AML-LSCs compared with normal HSCs. CD47 binds to the ligands thrombospondin-1 and SIRPα, which are involved in apoptosis, proliferation, adhesion, and migration of myeloma cells [24]. In clinical settings, increased expression of CD47 is a poor prognostic factor in AML. Treatment of human AML-engrafted mice with an anti-CD47 antibody could target AML-LSCs [25].
CD96 belongs to the Ig superfamily. Hosen et al. revealed that CD96 is a promising candidate as a specific antigen for LSCs [26]. Applying a signal sequence trap strategy to identify cell surface molecules expressed on human AML-LSCs and fluorescence-activated cell sorting (FACS) analysis demonstrated that CD96 is expressed on CD34+CD38− AML cells in the majority of patients. Transplantation of AML cells into immunodeficient mice demonstrated that only CD96+ fractions showed significant levels of engraftment. These results demonstrate that CD96 is a cell surface marker present on AML-LSCs and may be a LSC-specific therapeutic target. Furthermore, FACS analysis demonstrates that CD96 is expressed on the majority of CD34+CD38− LSCs of AML, whereas only a few normal HSCs weakly express CD96. Therefore, the authors mentioned that this molecule may be a candidate LSC-specific therapeutic target.
CD123 is an IL-3 receptor α chain, is highly expressed on AML-LSCs compared with their normal counterparts, and is being used to develop an antibody therapy. An anti-CD123-neutralizing antibody (7G3) targeted AML-LSCs, impairing homing to BM and activating innate immunity in NOD/SCID mice. 7G3 treatment profoundly reduced AML-LSC engraftment and improved mouse survival [27]. Targeting LSCs using the 7G3 antibody against CD123 seems to be an attractive approach. A phase 1 clinical study was conducted of relapsed or refractory high-risk AML using CSL360, a chimeric variant of 7G3; however, the remission rate was quite low [28]. One reason for this was the high tumor load, and strategies such as enhancing the ADCC (antibody-dependent cellular cytotoxicity) activity of anti-CD123 antibodies have been investigated. Moreover, it is an immune cell therapy, and preclinical work with a dual-affinity retargeting molecule generated from antibodies against CD3 and CD123, which is designed to redirect T cells against AML-LSCs, has been reported [29].
T-cell immunoglobulin mucin (TIM)-3 is highly expressed in AML-LSCs, except the FAB classification M3, but is not expressed at all in normal HSCs. While TIM-3-positive AML cells can repopulate AML in mice with high efficiency, TIM-3-negative AML cells do not reconstitute leukemia at all. In immunodeficient mice engrafted with AML cells and treated with an anti-TIM-3 antibody (ATIK2a), AML cells were significantly reduced, and those remaining after treatment with ATIK2a could not succeed in serial passage transplantation in mice, indicating that this antibody can target AML-LSCs [30]. TIM-3 protein is not detectable in normal HSCs or in other myeloerythroid or lymphoid progenitors, although monocyte lineage-committed progenitors begin to upregulate TIM-3. Therefore, expression of TIM-3 is highly specific to human AML-LSCs [31]. TIM-3 was originally identified as being selectively expressed on IFN-γ-secreting Th1 and Tc1 cells [32]. Interaction of TIM-3 with its ligand, galectin-9, triggers cell death in TIM-3+ T cells [33]. Both TIM-3 and PD-1 can function as negative regulators of T-cell responses [34, 35, 36]. Like PD-1, TIM-3 is also an immune checkpoint-associated surface molecule involved in the exhaustion of T cells. Inhibition of TIM-3 functions with an anti-TIM-3 antibody should be an effective treatment strategy. From the perspective of the abovementioned immune checkpoint inhibition, TIM-3 is expressed in some types of hematopoietic cells and seems to have lineage- or cellular context-dependent signal transduction pathways or functions. The role of TIM-3 in AML-LSCs is quite intriguing, and inhibition of this molecule is a promising clinical strategy. Further studies, however, are needed to clarify the specific function of TIM-3 before performing clinical trials to eradicate AML-LSCs.
IL1RAP is a co-receptor of the type 1 IL-1 receptor. IL1RAP is expressed on the cell surface in the majority of AML patients. IL1RAP is upregulated on immature cells in high-risk AML patients with chromosome 7 abnormalities, and AML patients with high IL1RAP expression have a poor prognosis. Ågerstam et al. reported that IL1RAP may become a therapeutic target in AML and that rapid clinical development of an antibody-based IL1RAP therapy for AML is promising [37].
CLL-1 is highly expressed on the blast compartment in the majority of AML cases, while it is completely absent on CD34+CD38− resting BM cells. CD34+CLL-1+ cells repopulate in sublethally irradiated immunodeficient NOD/SCID mice, indicating that they contain leukemia-initiating cells. Furthermore, culture of normal CD34+ cells in a long-term culture system in the presence of an anti-CLL-1 antibody has no effect on CFU-E and CFU-GM colony formation, suggesting that CLL-1 is a possible target for therapy [38].
20.4.3 Novel Strategy to Exhaust LSCs
The role of CCAAT/enhancer-binding protein β (C/EBPβ), a regulator of emergency granulopoiesis, in the pathogenesis of the chronic phase of CML was examined, and it has been suggested that C/EBPβ is involved in BCR-ABL-mediated myeloid expansion. We found that enforced expression of C/EBPβ in CML-LSCs might induce exhaustion of LSCs, leading to a complete cure of this disease [39]. C/EBPβ-mediated LSC loss might reveal a unique therapeutic strategy to eradicate CML stem cells. Agents that upregulate expression of C/EBPβ in LSCs should be promising (Yokota A, Hirai H, Maekawa T, et al., unpublished observation).
20.4.4 Targeting LSCs Using Small Molecules
In addition to therapies using the antibodies mentioned above, therapies based on small molecules can target intracellular proteins and pathways. Nuclear factor-kappa B (NF-κB) stimulates the transcription of genes encoding Ig of the κ class in B lymphocytes. NF-κB, a dimeric transcription factor complex that controls various aspects of cellular responses to stimuli, is now recognized as a pivotal regulator of cell survival, proliferation, and differentiation. When unstimulated, the complex is sequestered in the cytoplasm by a family of inhibitors of NF-κB (IκBs). NF-κB is constitutively active in AML enriched with LSCs, while its activity was not detectable in unstimulated normal HSCs/progenitor cells. Proteasome inhibitor such as MG-132, which inhibits NF-κB activity by inhibiting IκB degradation, induces the rapid apoptosis of AML-LSCs but not of normal HSCs. Therefore, inhibition of NF-κB signaling may provide a novel therapeutic strategy specific to LSCs [40]. Another molecule with inhibitory activity against NF-κB, parthenolide (PTL), targets primitive AML cells. Agents such as PTL, which is a sesquiterpene lactone that is the major active component in feverfew (Tanacetum parthenium), a herbal medicine that has been used to treat migraines and rheumatoid arthritis for a long time, inhibit the ability of LSCs to respond to oxidative stress and make LSCs sensitive to cell death stimuli, while normal stem cells remain relatively unharmed by these agents. The main mechanism of action of these molecules appears to revolve around the altered glutathione metabolism pathway found in leukemia cells [41]. Niclosamide also inhibits the transcription and DNA binding of NF-κB. It blocked tumor necrosis factor-induced IκBα phosphorylation, translocation of p65, and expression of NF-κB-regulated genes [42].
20.4.5 Targeting Antiapoptotic Molecules
A body of investigations has established that AML cells are dependent on antiapoptotic molecules for survival such as the B-cell CLL/lymphoma 2 (Bcl-2) family, which suppresses the intrinsic or mitochondrial apoptotic pathway and plays an important role in AML pathogenesis, prognosis, and responsiveness to chemotherapeutic agents [43]. In addition, the antiapoptotic Bcl-2 family protein myeloid cell leukemia sequence 1 (Mcl-1) is essential for cell survival during the development and maintenance of AML [44, 45].
Normally, the antiapoptotic proteins Bcl-2, Bcl-XL, and Mcl-1 suppress apoptosis effector molecules such as Bak as well as the proapoptotic BH3-only protein Bim, thereby preventing apoptosis. The BH3-only proteins Bad and Noxa, as well as BH3-mimetic drugs such as navitoclax and obatoclax, untether the apoptosis effectors and Bim from the antiapoptotic proteins, leading to apoptosis [46]. Apoptosis is initiated when members of the third subfamily, the BH3-only proteins (e.g., Bim, Bad, and Noxa), are activated by various cytotoxic stimuli.
20.5 Gene Abnormalities Involved in Epigenetic Modifications
Gene mutation analyses of leukemia cells using next-generation sequencing greatly assist studies of individual genetic abnormalities in leukemia cells. In addition, the importance of methylation of DNA and histones for epigenetic regulation of gene expression by modulation of the chromatin structure was recently clarified. Abnormalities of genes related to epigenetic modifications are frequently found in many cancers, including AML. There are roughly two types of epigenetic modifications; one is histone modifications including the polycomb group complex-associated gene ASXL1, and the other is DNA methylation modifications including the DNA methyltransferase 3A (DNMT3A), 10–11 translocation 2 (TET2), isocitrate dehydrogenase 1 (IDH1), and IDH2 genes [47, 48]. Gene expression can be controlled by these chemical modifications without changing the genome sequence.
Somatic mutations in genes that encode proteins related to epigenetic alterations play a pivotal role in leukemogenesis. Shlush et al. recently reported that mutations of the DNMT3A, TET2, and IDH1/2 genes, which control DNA methylation, and the polycomb group complex-associated gene ASXL1, which is involved in histone modifications, are involved in the initiation of pre-LSCs as driver mutations. Such mutations facilitate the self-renewal capacity of pre-LSCs and block the differentiation of hematopoietic stem/progenitor cells. The emergence of these mutations in HSCs can induce their clonal expansion, resulting in a pre-LSC population. Furthermore, HSCs with this DNMT3A gene mutation were demonstrated to have a growth advantage in a patient’s BM using a xenograft model. Similar findings were observed in HSCs with ISH2 gene mutation; therefore, DNMT3A and IDH2 gene mutations have a critical role in the preleukemic state [49]. Hemopoietic cells with these gene mutations can remain after a long period of remission achieved by chemotherapy or molecular targeted therapy, and these cells may cause leukemia relapse [50]. The development of a histone deacetylase inhibitor is underway in clinical settings to target such abnormal molecules. AR-42 (OSU-HDAC42) induces NF-κB inhibition, disrupts the ability of heat shock protein 90 to stabilize its oncogenic clients, and causes potent and specific apoptosis of LSCs but not of normal hematopoietic stem and progenitor cells. AR-42 has already been tested in early clinical settings [51].
DNA is methylated at the 5-position carbon in CpG sequences. Methylated cytosine-binding proteins bind to the methylated cytosine, and gene transcription is suppressed together with recruited histone deacetylase and transcriptional repressor. Regions where CpG sequences are condensed are called CpG islands. CpG islands are quite abundant in the promoter region of a gene. CpG islands are usually maintained in a low methylated condition, and gene transcription is activated. In cancer, genome-wide hypomethylation is frequently found, and a specific promoter region is hypermethylated. Genome-wide hypomethylation results in chromosomal instability, along with reactivation of the transposon, which is thought to be the cause of genetic abnormalities. On the other hand, hypermethylation of DNA in CpG islands suppresses the expression of CDK inhibitors, which are negative regulators of the cell cycle and inhibit the expression of tumor suppressor genes and DNA repair genes. Such an altered methylated status has become a therapeutic target, and the development of DNA methylation inhibitors and histone deacetylase inhibitors is under investigation. The detailed mechanism of action in hyper- and hypomethylation will be discussed elsewhere in this book.
20.6 Targeting the Hypoxic Niche of LSCs and Their Interactions
To maintain their undifferentiated state, LSCs, similar to HSCs, adhere to the stem cell niche of the BM microenvironment, which is composed of a variety of cell types, including mesenchymal stem/stromal cells, bone cells, immune cells, neuronal cells, and vascular cells [52]. It is possible that LSCs obtain anticancer drug resistance by lodging in the niche and that the interactions that maintain LSCs are the major cause of leukemia relapse. Angiopoietin-1, Flt3 ligand, thrombopoietin, Wnt/β-catenin [16], CXCL12/SDF-1α [53], CD44, addition of osteopontin, basic fibroblast growth factor, insulin-like growth factor, IL-6, vascular endothelial growth factor, and many other molecules are involved in stem cell maintenance in the niche. By blocking the interaction between LSCs and niches, it is possible to induce quiescent LSCs in the niche to enter the cell cycle, and then these quiescent LSCs could become a target of therapeutic molecules such as chemotherapeutic agents and various inhibitors. HSCs utilize the system of chemokine receptor-4 (CXCR4) and CXCL12/SDF-1α binding [54]. When AML cells were implanted into mice, more engraftment was observed in cases highly expressing CXCR4, and CXCR4/SDF-1α signaling promotes the survival of leukemia cells by inhibiting their differentiation. An anti-CXCR4 agent inhibits the binding of AML-LSCs in the niche, mobilizing AML-LSCs from the niche to the periphery, which is expected to increase sensitivity to chemotherapy [55, 56].
Related posts:
 Bcr-Abl Tyrosine Kinase Inhibitors (TKIs)
Inotuzumab Ozogamicin for Acute Lymphoblastic Leukemia: Clinical Pharmacology and Therapeutic Results
Bcr-Abl Tyrosine Kinase Inhibitors (TKIs)
Inotuzumab Ozogamicin for Acute Lymphoblastic Leukemia: Clinical Pharmacology and Therapeutic Results
 Nelarabine
Nelarabine
 Rituximab and Alemtuzumab for Chronic Lymphocytic Leukemia: Basic Results and Pharmacokinetics
Rituximab and Alemtuzumab for Chronic Lymphocytic Leukemia: Basic Results and Pharmacokinetics
 Imatinib: Basic Results
Imatinib: Basic Results
 Imatinib: Clinical Pharmacology and Therapeutic Results
Imatinib: Clinical Pharmacology and Therapeutic Results
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


