Therapeutics in older people
Stephen Jackson
Key points
 Pharmacokinetics in older people is different to that in younger people:
Pharmacokinetics in older people is different to that in younger people:
• renal clearance is lower (water-soluble drugs)
• hepatic clearance is lower (lipid-soluble drugs)
• half-life is further prolonged for lipid-soluble drugs because of the increased volume of distribution of such drugs
• in frail older patients, half-life is further prolonged
• by reduced hepatic enzyme activity (lipid-soluble drugs)
• by reduced protein binding and hence increasing the volume of distribution (very heavily protein-bound drugs).
 Polypharmacy is common and reflects multiple pathology.
Polypharmacy is common and reflects multiple pathology.
 Inappropriate medication should always be avoided.
Inappropriate medication should always be avoided.
 Methods of enhancing the quality of prescribing include
Methods of enhancing the quality of prescribing include
• regular medication review
• prescribing audit using proven indicators of appropriateness
• education of prescribers.
1 Introduction
The size of the oldest old section of the population is rising and is set to continue to increase (1), Chapter 1). This population has a higher prevalence of chronic illnesses including cardiovascular diseases, cancers, osteoporosis, diabetes, Parkinson’s disease, dementia, and many other conditions. An increasing body of research is adding further prescribing indications for diseases that occur in the elderly population. Along with the increasing size of the population, this means that the numbers of prescriptions for elderly patients are increasing. Depending on the age group, between 60% and 80% of elderly people are taking medication and 20% are taking at least five drugs (2). It is also estimated that although those over 75 years account for 14% of the population, they receive 33% of medication prescribed.
2 Age-related changes in physiology and pharmacokinetics
Pharmacokinetics is the description of how the body handles drugs after administration. It incorporates the liberation, absorption, distribution, metabolism, and excretion of drugs and their metabolites. These processes are affected by the physiological changes associated with ageing resulting in changes in the pharmacokinetics of drugs.
2.1 Absorption
Following administration, a number of factors affect a drug’s entry into the circulation. These include properties such as particle size, molecular weight, charge, solubility, and pKa (the pH at which 50% remains in a non-ionized, lipid-soluble state). Most drugs are weak acids or bases and are present in solution as both ionized and non-ionized species. Non-ionized drugs are lipid-soluble and diffuse easily across the cell membrane. Following liberation some absorption may take place in the stomach, depending on the pKa of the drug and the pH of the stomach. Gastric acid secretion in response to stimulation decreases with normal ageing. Thus where pH is crucial to ionization, absorption can be affected (e.g. iron compounds). However, for most drugs the large surface area of the small bowel makes it the main site of drug absorption.
With ageing, although many drugs show no change in absorption (3), there is slightly reduced small bowel absorption of some substances (including iron, calcium, and glucose). There is slower colonic transit time with age, and an associated decrease in peristalsis, largely due to a loss of neurons involved in control of the GI tract. Passive intestinal permeability is probably unchanged in old age for most substances. However, active transport of other agents, such as vitamin B12, is impaired. These age-related changes therefore primarily affect drugs with low permeability and low solubility, e.g. cephalexin. Agents such as benzodiazepines (lipid-soluble) and lithium (water-soluble) are readily absorbed.
2.2 Distribution
The volume of distribution (Vd), also known as apparent volume of distribution, is a pharmacological term used to quantify the distribution of a medication between plasma and the rest of the body after oral or parenteral dosing. It is defined as the volume in which an amount of drug would need to be uniformly distributed to produce a given plasma concentration. Put another way, it refers to the fluid volume that would be required to contain the entire drug dose in the body at the same concentration as that in the blood or plasma. A drug with a high Vd (e.g. morphine—300 litres) implies extensive distribution outside the blood or plasma to other tissues such as fat. The Vd is dependent on lipophilicity (increasing the Vd) and the ability of the drug to bind to plasma proteins such as albumin (acidic drugs) and α1-acid glycoprotein (basic drugs), thus holding the drug in the blood compartment and reducing Vd.
With ageing there is a decrease in lean body weight, muscle mass, and body water, and an increase in body fat per kilogram of body weight. As a result, lipid-soluble drugs such as benzodiazepines, morphine, neuroleptics, and amitriptyline have an increased Vd due to the higher proportion of body fat. With ageing the higher Vd for such lipid-soluble drugs (along with reduced clearance) will result in a prolonged elimination half-life and hence drug effects. For water-soluble drugs, Vd will fall, but clearance will fall to a greater extent.
2.3 Protein binding
Many drugs are protein-bound to a varying degree. Bound drugs are inactive. An unbound drug is free to mediate its effect. The binding is usually reversible. Most acidic drugs (e.g. ibuprofen, diazepam, phenytoin, warfarin) bind to albumin. Basic drugs such as lidocaine and tricyclic antidepressants bind to α1-acid glycoprotein. With healthy ageing there is no substantial change in plasma proteins; however, intercurrent illness can result in a drop in albumin and an increase in α1-acid glycoprotein (an acute phase protein). Chronic disease tends to accelerate the age-related decline in serum albumin. This can produce clinically significant increases in the free fraction of very heavily protein-bound drugs such as ibuprofen (99.5% bound). Other highly protein-bound drugs include benzodiazepines (>90%) and many antipsychotics (>90%). In contrast, some drugs are not protein-bound at all (e.g. lithium).
2.4 Clearance—hepatic
Hepatic metabolism of drugs is dependent on the ability of the liver to extract drugs from the blood passing through the organ. Lipophilic drugs are metabolized into more hydrophilic compounds that are eliminated mainly through the urine. However, in some cases metabolites are biologically active or even toxic. Thus risperidone is metabolized to an active metabolite (9-OH risperidone), which has an elimination half-life (t1/2z) of 22 hours versus the parent drug t1/2z of 4 hours. Similarly, diazepam is metabolized to an active metabolite (desmethyl diazepam), amitriptyline is metabolized to nortriptyline, and morphine is metabolized to the active metabolite morphine-6-glucuronide. In the liver, phase I metabolism introduces a functional group onto the parent compound, generally resulting in loss of pharmacological activity. Several studies have shown an age-related decline in the clearance of drugs by phase I metabolism, probably reflecting a reduced hepatic mass as enzyme activity is preserved. Induction, or increased synthesis of the cytochrome P450 enzymes induced by drugs such as phenytoin, isoniazid, glucocorticoids, and alcohol, may decrease the bioavailability of parent drug compounds.
Inhibition of drug metabolism enzymes, commonly by depletion of necessary co-factors, results in elevated levels of parent drugs. This can lead to increased pharmacological effects and an increased incidence of drug-induced toxicity. Inhibition of different isoforms of cytochrome P450 enzymes can be seen with erythromycin and ketoconazole (CYP450 3A4) and SSRIs (CYP450 2D6). For two of these enzymes, CYP4502D6 and CYP4502C19, genetic polymorphisms exist that lead to poorly functioning enzymes causing individuals to be poor metabolizers. Thus when prescribing drugs metabolized by these enzymes where the therapeutic window is narrow, prescribing should be on the basis that the patient is a poor metabolizer. For example, when prescribing haloperidol, starting doses should be appropriately low in poor CYP2D6 metabolizers.
2.5 Clearance—renal
Excretion of drugs and metabolites in the urine involves three processes: glomerular filtration, active tubular secretion, and passive tubular reabsorption. With ageing, renal mass decreases, as does the glomerular filtration rate (GFR). There is also a reduced ability to concentrate urine and a reduced thirst during water deprivation.
Davies and Shock, in a classic cross-sectional inulin clearance study, demonstrated that GFR decreases by about 8ml/min/1.73 m2 per decade from the fourth decade onwards (4). There is wide individual variability in the age-related fall in GFR, further amplified by the presence of vascular and renal disease. Creatinine clearance is influenced by nutritional status, protein intake, muscle mass, body weight, gender, and ethnicity. As people age, muscle mass is reduced and daily urinary creatinine excretion decreases, accompanied by a reduction in creatinine clearance. The combined effect of these changes is that declining GFR in older patients is accompanied by lower rises in serum creatinine than would occur in younger people.
Reduction in GFR with age affects the clearance of many drugs such as water-soluble antibiotics, diuretics, lithium, and water-soluble non-steroidal anti-inflammatory drugs. GFR can be estimated using several equations. The Cockcroft and Gault equation uses age, weight, gender, and serum creatinine (5):
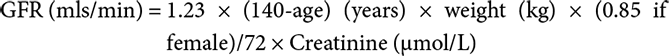
The National Service Framework for Kidney Disease recommended that all laboratories report a formula-based estimation of GFR when serum creatinine is requested in adults. The Modification of Diet in Renal Disease (MDRD) study equation (based on serum creatinine, age, gender, and ethnic group) (6) is widely used. The MDRD formula has the advantage of not requiring a weight, and can therefore be issued by the laboratory at the same time as a creatinine result is reported. It takes no direct account of muscle mass. It estimates the reduction in muscle bulk on the basis of the average reduction due to age. The classification of chronic kidney disease (Box 5.1) is based on eGFR estimated by the MDRD formula. The Cockcroft and Gault formula tends to estimate lower values for GFR than MDRD estimates. Some data suggest that the MDRD formula is unreliable in end-stage renal disease (7).
Box 5.1 Stages of chronic kidney disease (CKD)
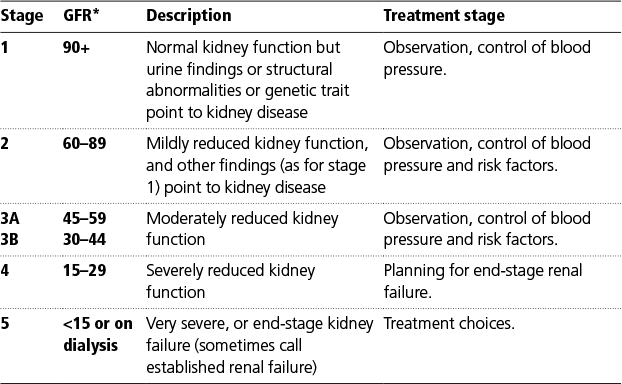
* All GFR (glomerular filtration rate) values are normalized to an average body surface area (size) of 1.73m2
Acknowledgement: The Renal Association—original authors Goddard J, Harris K, and Turner N. <http://www.renal.org/information-resources/the-uk-eckd-guide/ckd-stages#sthash.yN5edpAX.orJDyUxb.dpbs>
Related posts:
 Incontinence, the sleeping geriatric giant: challenges and solutions
Health and social care services for older people: achievements, challenges, and future directions
Substance misuse and older people: a question of values
Service models
Dementia and memory clinics
Assessment and management of pain in older adults
Incontinence, the sleeping geriatric giant: challenges and solutions
Health and social care services for older people: achievements, challenges, and future directions
Substance misuse and older people: a question of values
Service models
Dementia and memory clinics
Assessment and management of pain in older adults
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


