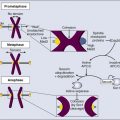
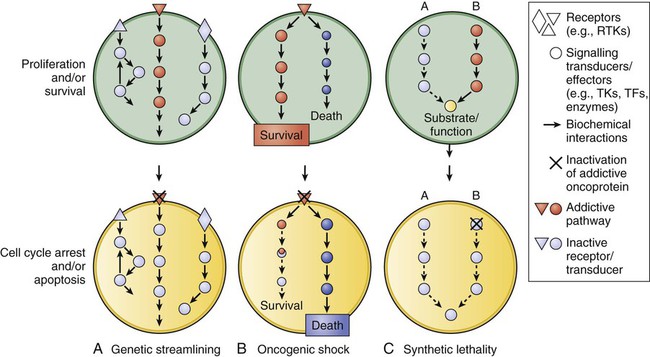
Shivaani Kummar, Anthony J. Murgo, Joseph E. Tomaszewski and James H. Doroshow • Molecularly targeted anticancer agents (MTAs) are those that selectively target specific molecular features of cancer cells such as aberrations in genes, proteins, or pathways that regulate tumor growth, progression, and survival. • Molecular targets include the following: products of activating mutations and translocations, growth factors and receptors, aberrant signal transduction and apoptotic pathways, factors that control tumor angiogenesis and microenvironment, dysregulated proteins, DNA repair machinery, and aberrant epigenetic mechanisms. • The development of MTAs requires innovative strategies that differ from those traditionally applied to nontargeted conventional chemotherapy. • Successful development of an MTA depends largely on the importance of the target in controlling tumor cell proliferation and survival and effective modulation of the target in the tumor at clinically achievable concentrations. • Primary objectives of clinical trials of MTAs differ from those used in trials of conventional chemotherapy. An important objective in phase 1 trials of MTAs should be to determine a phase 2 dose based on optimal target modulation (i.e., a biologically effective dose) rather than on maximum tolerated dose. In addition, objective tumor response may not be an adequate end point for efficacy evaluations of MTAs that have a primarily cytostatic effect. Alternate end points, such as progression-free survival, may be more appropriate. • Functional and molecular imaging plays an increasingly important role in the development of MTAs. The increasing number and assortment of molecular targets can be broadly categorized according to genetic or functional properties, including products of activating gene mutations and translocations; growth factors and receptors; aberrant signal transduction and apoptotic pathways; factors that control tumor angiogenesis and microenvironment; dysregulated proteins; DNA repair machinery; and aberrant epigenetic mechanisms (Tables 28-1 and 28-2). Table 28-1 U.S. Food and Drug Administration–Approved Molecularly Targeted Agents Table 28-2 Classes of Molecularly Targeted Anticancer Agents under Clinical Development The most promising molecular targets are those solely responsible for sustaining tumor growth and survival because agents that potently and selectively inhibit these critical targets are likely to have a major clinical impact. Probably the best example of a critical target is BCR-ABL in patients with chronic myelogenous leukemia (CML). BCR-ABL is a fusion protein formed by the reciprocal translocation of chromosomes 9 and 22. Knowledge that this dysregulated tyrosine kinase played a causal role in the pathogenesis of essentially all cases of CML spurred preclinical studies, which led to the development of a potent and selective ABL tyrosine kinase inhibitor (TKI), imatinib mesylate (previously known as STI571).1 Subsequent clinical trials established imatinib mesylate as the first highly effective molecularly targeted therapy for CML and a prototype for the development of others in the class. Imatinib mesylate is also a potent inhibitor of other tyrosine kinases including PDGFR and KIT, and it is highly effective in the treatment of gastrointestinal stromal tumors (GISTs) bearing activating c-KIT mutations and in some GISTs bearing activating PDGFR mutations.1,2 Unfortunately, most human tumors, including the most common types, are genetically complex and do not have a single critical target, and the relative critical importance of a target in different tumors may vary. Most tumor types have various genetic and molecular abnormalities driving their growth and survival. The existence of multiple abnormalities in one or more molecular pathways helps explain resistance to molecularly targeted therapy and provides a rationale for treatment strategies combining two or more targeted agents.3,4 However, cancer cells may become “addicted” or physiologically dependent on the sustained activity of specific oncogenes for maintenance of a malignant phenotype and for survival. This dependence mechanism, termed oncogene addiction7–7 (Fig. 28-1), is associated with differential attenuation rates of prosurvival and proapoptotic signals stemming from the oncoprotein, with predominant apoptotic signals resulting in cell killing. The latter process, termed “oncogenic shock,”8 could explain the remarkably rapid clinical responses to TKIs in some patients with solid tumors, including those typically having complex molecular abnormalities. Other possible factors controlling sensitivity or resistance to molecularly targeted therapy include increased expression of the target due to gene amplification or transcription, emergence of resistant target gene mutations, and overexpression of multidrug transporter membrane proteins.4,7 Interest in molecular therapy directed at factors controlling angiogenesis has increased since the United States Food and Drug Administration (FDA) approved several agents that target vascular endothelial growth factor (VEGF) and its receptor (VEGFR). The VEGF pathway, involving the VEGF family of proteins and their receptors, is an important regulator of both physiological and pathological angiogenesis. Through its signaling pathways, VEGF/VEGFR activation contributes to increased vascular permeability, mobilization of bone marrow–derived endothelial cell precursors, degradation of the extracellular matrix, and endothelial cell division, differentiation, migration, and survival.9,10 VEGF overexpression occurs in most types of cancers, including colorectal, renal, gastric, pancreatic, liver, lung, breast, thyroid, and genitourinary; it also occurs in glioma and other intracranial tumors, as well as in hematologic malignancies, and it is associated with tumor growth and a worse clinical outcome in a number of these tumor types.10,11 Potential therapeutic strategies to inhibit signaling through VEGF and VEGFR pathway activation include monoclonal antibodies directed against VEGF or VEGFR, TKIs, and antisense strategies (antisense oligodeoxynucleotides, antisense RNA, and small interfering RNAs). In 2004, the FDA approved bevacizumab, a humanized murine monoclonal antibody directed against VEGF, for treating metastatic colorectal cancer in combination with fluorouracil-based chemotherapy. The FDA subsequently approved sunitinib, sorafenib, and axitinib, three small-molecule TKIs with activity against VEGFR, for the treatment of advanced renal cancer.11 In addition, sorafenib is effective for treating hepatocellular carcinoma, a tumor against which standard cytotoxic chemotherapy has little or no activity.11,12 The experience with these agents has established the VEGF/VEGFR pathway as a valid target for cancer therapeutics.11 Mammalian target of rapamycin (mTOR) has also emerged as a validated target with the demonstration that the small-molecule mammalian target of rapamycin inhibitors temsirolimus and everolimus are effective in the treatment of renal cell carcinoma.13 This activity of temsirolimus is attributed to the downregulation of factors that control cell growth and angiogenesis.13 The discovery and development of molecularly targeted therapies requires closely aligned laboratory and clinical research, integrating drug discovery, development, and clinical investigation. In such a cooperative setting, researchers can effectively take rational and iterative steps from target identification to clinical evaluation (Box 28-1) (Fig. 28-2). A crucial early step in developing a molecularly targeted therapy is target validation, defined as experimental evaluation of the role of a given gene or protein.14,15 The target validation process involves a variety of preclinical approaches, including genetic, cell-based, and animal models.16 Validation and prioritization of molecular targets for therapeutic development depends on a variety of criteria, taking into consideration chemical, biological, clinical, and practical factors15 (Box 28-2). The fundamental goal is to provide evidence that the target is valid (i.e., that affecting the target inhibits tumor growth, progression, or survival) and that making drugs that hit the target is feasible. The next major step in the development of molecularly targeted therapy is finding/synthesizing compounds directed against that target. Box 28-2 Validation Criteria and Prioritization of New Targets for Drug Screening • High frequency of genetic or epigenetic deregulation of the molecular target or pathway in human cancer indicates that the target or pathway is likely important in driving the disease • Linkage of the deregulation to clinical outcome strengthens the case for causal involvement • Evidence in a model system that the target pathway causes or contributes to the malignant phenotype demonstrates a direct causal role in malignancy • Demonstration of reversal of the malignant phenotype provides greater confidence that modulation of the target by a drug will produce an anticancer effect • Demonstration of “drugability” of the target—for example, enzymes are generally much more “drugable” than are large-domain protein-protein interactions • Availability of a robust, efficient biological test cascade to support the drug discovery program to allow evaluation of lead compounds and to select a development candidate for preclinical toxicology testing and clinical trials • Feasibility of establishing, validating, and running an affordable and robust high-throughput screen
Therapeutic Targeting of Cancer Cells
Era of Molecularly Targeted Agents
Molecular Targets
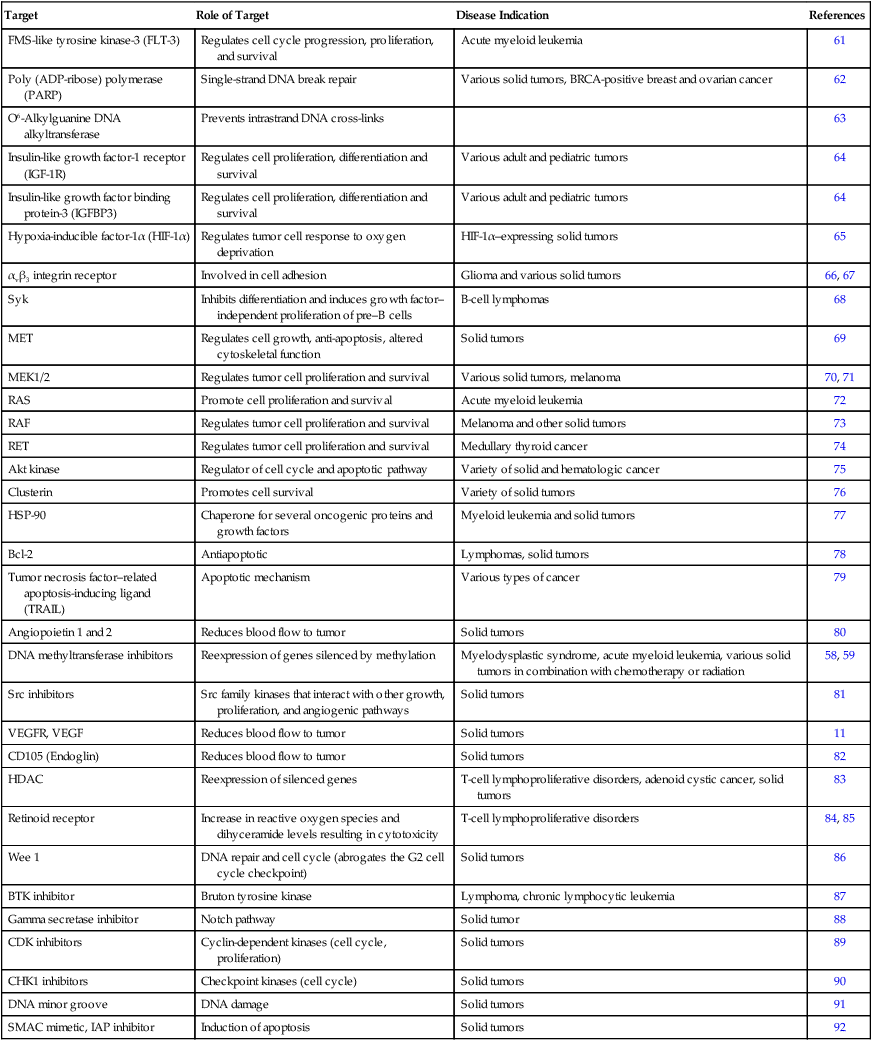
FDA Approved Agents(s)
Target
Disease Indication
Imatinib mesylate
BCR-ABL, KIT, PDGFR-β
Ph+ CML, Ph+ ALL, chronic monomyelocytic leukemia, dermatofibrosarcoma protuberans
Dasatinib
BCR-ABL, Src family kinases
Ph+ CML
Nilotinib hydrochloride monohydrate
BCR-ABL
Ph+ CML
All-trans retinoic acid
PML-RAR
Acute promyelocytic leukemia
Sunitinib malate
VEGFR, KIT, PDGFR-α
GIST, pancreatic neuroendocrine tumors, kidney cancer
Vemurafenib
BRAFV600E mutation
BRAFV600E-positive melanoma
Crizotinib
ALK tyrosine kinase
ALK-positive non–small cell lung cancer
Erlotinib
EGFR
Non–small cell lung cancer
Panitumumab
EGFR
Colorectal cancer
Cetuximab
EGFR
Head and neck cancer, colorectal cancer
Trastuzumab
ErbB-2
HER2 overexpressing breast cancer
Lapatinib
ErbB-2
HER2 overexpressing breast cancer
Vismodegib
Hedgehog pathway
Advanced/metastatic basal cell carcinoma
Brentuximab vedotin
CD30
Hodgkin lymphoma, anaplastic large cell lymphoma
Ofatumumab
CD20
CLL
Ibritumomab
Non-Hodgkin lymphoma
Rituximab
B-cell non-Hodgkin lymphoma
Alemtuzumab
CD52
B-cell chronic lymphocytic leukemia
Bevacizumab
VEGF
Colorectal cancer, non–small cell lung cancer, renal cell carcinoma
Vandetanib
VEGFR
Medullary thyroid cancer
Sorafenib
VEGFR, B-Raf kinase
Kidney cancer, hepatocellular cancer
Pazopanib
VEGFR
Soft tissue sarcomas, kidney cancer
Axitinib
VEGFR
Kidney cancer
Temsirolimus
mTOR
Renal cell carcinoma
Everolimus
mTOR
Renal cell carcinoma, advanced pancreatic neuroendocrine tumors
Azacitidine
DNA methyltransferase
Myelodysplastic syndrome
Decitabine
DNA methyltransferase
Myelodysplastic syndrome
Vorinostat
Histone deacetylase
Cutaneous T-cell lymphoma
Romidepsin
Histone deacetylase
Cutaneous T-cell lymphoma
Bortezomib
Proteosome
Multiple myeloma
Ipilimumab
Cytotoxic T-lymphocyte–associated antigen 4
Metastatic melanoma
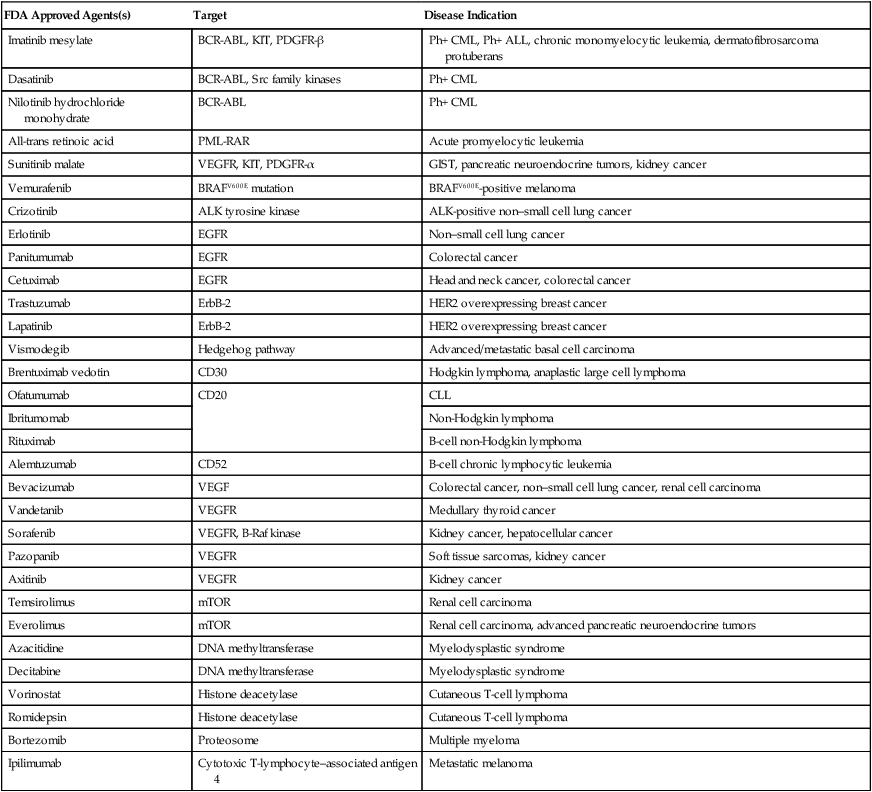
Target
Role of Target
Disease Indication
References
FMS-like tyrosine kinase-3 (FLT-3)
Regulates cell cycle progression, proliferation, and survival
Acute myeloid leukemia
61
Poly (ADP-ribose) polymerase (PARP)
Single-strand DNA break repair
Various solid tumors, BRCA-positive breast and ovarian cancer
62
O6-Alkylguanine DNA alkyltransferase
Prevents intrastrand DNA cross-links
63
Insulin-like growth factor-1 receptor (IGF-1R)
Regulates cell proliferation, differentiation and survival
Various adult and pediatric tumors
64
Insulin-like growth factor binding protein-3 (IGFBP3)
Regulates cell proliferation, differentiation and survival
Various adult and pediatric tumors
64
Hypoxia-inducible factor-1α (HIF-1α)
Regulates tumor cell response to oxygen deprivation
HIF-1α–expressing solid tumors
65
αvβ3 integrin receptor
Involved in cell adhesion
Glioma and various solid tumors
66, 67
Syk
Inhibits differentiation and induces growth factor–independent proliferation of pre–B cells
B-cell lymphomas
68
MET
Regulates cell growth, anti-apoptosis, altered cytoskeletal function
Solid tumors
69
MEK1/2
Regulates tumor cell proliferation and survival
Various solid tumors, melanoma
70, 71
RAS
Promote cell proliferation and survival
Acute myeloid leukemia
72
RAF
Regulates tumor cell proliferation and survival
Melanoma and other solid tumors
73
RET
Regulates tumor cell proliferation and survival
Medullary thyroid cancer
74
Akt kinase
Regulator of cell cycle and apoptotic pathway
Variety of solid and hematologic cancer
75
Clusterin
Promotes cell survival
Variety of solid tumors
76
HSP-90
Chaperone for several oncogenic proteins and growth factors
Myeloid leukemia and solid tumors
77
Bcl-2
Antiapoptotic
Lymphomas, solid tumors
78
Tumor necrosis factor–related apoptosis-inducing ligand (TRAIL)
Apoptotic mechanism
Various types of cancer
79
Angiopoietin 1 and 2
Reduces blood flow to tumor
Solid tumors
80
DNA methyltransferase inhibitors
Reexpression of genes silenced by methylation
Myelodysplastic syndrome, acute myeloid leukemia, various solid tumors in combination with chemotherapy or radiation
58, 59
Src inhibitors
Src family kinases that interact with other growth, proliferation, and angiogenic pathways
Solid tumors
81
VEGFR, VEGF
Reduces blood flow to tumor
Solid tumors
11
CD105 (Endoglin)
Reduces blood flow to tumor
Solid tumors
82
HDAC
Reexpression of silenced genes
T-cell lymphoproliferative disorders, adenoid cystic cancer, solid tumors
83
Retinoid receptor
Increase in reactive oxygen species and dihyceramide levels resulting in cytotoxicity
T-cell lymphoproliferative disorders
84, 85
Wee 1
DNA repair and cell cycle (abrogates the G2 cell cycle checkpoint)
Solid tumors
86
BTK inhibitor
Bruton tyrosine kinase
Lymphoma, chronic lymphocytic leukemia
87
Gamma secretase inhibitor
Notch pathway
Solid tumor
88
CDK inhibitors
Cyclin-dependent kinases (cell cycle, proliferation)
Solid tumors
89
CHK1 inhibitors
Checkpoint kinases (cell cycle)
Solid tumors
90
DNA minor groove
DNA damage
Solid tumors
91
SMAC mimetic, IAP inhibitor
Induction of apoptosis
Solid tumors
92
Preclinical Development of MTAs
Related posts:
![]()
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


Therapeutic Targeting of Cancer Cells: Era of Molecularly Targeted Agents