Keywords
IL-12, adenovirus, RheoSwitch®, activator ligand, dendritic cells, controlled expression, intratumoral, immunotherapy
Introduction
Tumors grow aggressively, are invasive, and may spread to distant metastatic sites, not only because of their intrinsic properties but also because they have developed a variety of mechanisms that alter the tumor microenvironment and evade the powerful potential assault of the immune system (reviewed in Hanahan and Weinberg ). The cancer immunoediting process provides a foundation for understanding the complex role of the immune system during tumor development , but it also allows for developing strategies to augment effective antitumor immunity, to inhibit tumor growth and spread, and even in some cases to induce complete tumor regression. Since the initial utilization of Coley’s toxin (reviewed in Thotathil and Jameson ), much more sophisticated immunotherapy approaches have been developed based on specific, well-defined targets and effector molecules, such as cytokines, and effector cells, such as T cells and/or natural killer (NK) cells. Recently, there has been accelerating progress in immunotherapy of cancer, not only in rodent tumor models but also in patients with cancer. However, the translation of effective immunotherapy strategies has often been hampered by toxicities and even mortality in some patients. For example, the use of some cytokines, administered systemically, has been approved by the U.S. Food and Drug Administration, including high-dose interleukin (IL)-2 (aldesleukin, Proleukin) and interferon (IFN)-α (Intron-A, IFN-α2b; Roferon-A, IFN-α2a); however, these have not been widely adopted by clinical oncologists because of serious toxicities and only limited therapeutic efficacy.
The identification in the early 1990s of NK-stimulating factor, now called IL-12 , a heterodimer (IL-12p70) consisting of disulfide-linked p35 and p40 chains, and its subsequent characterization have led to understanding of its importance in antitumor activity against tumors, infections, and autoimmunity . IL-12 has become recognized as a cytokine that bridges the actions of innate resistance with adaptive immunity . Furthermore, the intricate roles of the other members of the IL-12 heterodimeric family (IL-23, IL-27, and IL-35) have provided additional insights into resistance against cancer or infections and the development of autoimmunity . IL-12 is produced by antigen-presenting cells (APCs), such as subpopulations of macrophages and dendritic cells (DCs), following stimulation through the activation of the pattern recognition receptors such as toll-like receptors (TLRs) by pathogen-associated molecular pattern (PAMP) and cell stress-related danger-associated molecular pattern (DAMP) molecules . IL-12 binds to the IL-12 receptor, which is composed of β 1 and β 2 chains, and is expressed on NK cells and T cells and induces cell signaling via the JAK-STAT pathway. This signaling pathway results in activation of NK cells and T cells, differentiation of naive CD4 + T cells to a T helper 1 (Th1) phenotype, and differentiation of naive CD8 + T cells to cytotoxic T lymphocytes (CTLs) . It has also been shown that IL-12, either directly or indirectly, can enhance the maturation of APCs and can play an important role in the generation of antitumor immune responses . Central to the biological activities of IL-12 is its induction of the downstream cytokine IFN-γ produced by NK and T cells . Increased expression of IFN-γ can enhance the expression of major histocompatibility complex (MHC) antigens on tumor cells and APCs such as DCs, which in turn augments stimulation of naive T cells and differentiation into CTLs. IL-12 has also been shown to have anti-angiogenic effects, mediated by downstream cytokines such as IFN-γ, IP-10, and MIG, providing an additional mechanism of action to effectively eliminate tumor cells.
Systemic or Intratumoral Administration of IL-12
The early days of IL-12-based immunotherapy relied mainly on systemic administration of recombinant murine IL-12 (mIL-12), via daily dosing intraperitoneally, with highly encouraging antitumor and antimetastatic results in various mouse tumor models. These studies indicated that IL-12 as a single agent has the ability to delay or reduce tumor growth and even cause tumor regression. In addition, several groups have also established that the combination of IL-12 with other cytokines, such as IL-2, IL-18, IL-15, granulocyte–macrophage colony-stimulating factor (GM-CSF), or IFN-α, can further augment antitumor activity, either synergistically or additively, compared to either single cytokine treatment alone (as reviewed by Weiss et al. ).
Phase I and phase II clinical trials involving systemic delivery of recombinant human IL-12 (hIL-12) were performed in the mid- to late 1990s, and a few in the early 2000s, in patients with advanced solid tumors, such as renal cell carcinoma, melanoma, bladder cancer, head and neck cancers, and hematologic malignancies such as cutaneous T cell lymphoma (CTCL) . In phase I dose-escalation studies, using bolus IL-12 intravenously (i.v.), the maximum tolerated dose (MTD) was 500 ng/kg/day . As expected, the administration of IL-12 systemically resulted in induction of immune responses in some subjects, with stable disease in some patients seen at or above the established MTD . However, at these high i.v. doses, serious adverse events (SAEs) occurred, which included increases in liver transaminases, cytopenia, and even deaths . In a phase II trial evaluating repeated i.v. administration of hIL-12 in patients with renal cell carcinoma, 12 of 17 patients were hospitalized and 2 died due to hypovolemic shock . The study was halted because most patients experienced SAEs during the first cycle of treatment. There was a high frequency of grade 3 or 4 AEs, with 65% of patients developing leukopenia and 47% having hyperbilirubinemia and elevated AST levels . The clinical responses seen in this and other trials were infrequent, with only 4 of 48 (8.3%) or 1 of 29 (3.4%) showing complete, partial, or stable disease among evaluable patients in a phase II multiple myeloma trial. As an alternative to i.v. administration, some trials have administered hIL-12 subcutaneously (s.c.). The MTDs for s.c. administration were between 500 ng/kg and 1500 ng/kg , depending on the dosing schedule. These trials showed similar dose-limiting toxicities and minimal clinical benefit as after i.v. administration of IL-12.
In contrast to these reports from trials with systemic administration, therapy by the local delivery of IL-12 into the tumor may limit the systemic toxic effects from the induced cytokine cascade and be therapeutically more effective if it induces systemic as well as local antitumor effects. Clinical studies performed in patients with different forms of cutaneous T cell lymphoma, such as Sézary syndrome or mycosis fungoides, demonstrated beneficial clinical responses after s.c. delivery of hIL-12 at doses of 50–300 ng/kg . Treatments were well tolerated, although flu-like symptoms as well as injection site reactions were commonly reported. In one study, s.c. delivery achieved an overall response rate of 56% (including complete responders and partial responders) in patients with plaques and Sézary syndrome . Furthermore, it was demonstrated that intralesional injection with recombinant hIL-12 resulted in flattening and resolution of the injected lesions; however, the patients continued to develop additional lesions and progressed. Immunohistochemistry analysis from biopsy samples demonstrated an increased infiltration of CD3 + T cells but, more important, an increase in the number of CD8 + T cells, which are potentially tumor-reactive CTLs. In a phase II study, it was demonstrated that s.c. administration of IL-12 to 23 patients with mycosis fungoides resulted in 43% partial responders, 30% with minor response, and 22% with stable disease . Intratumoral (i.t.) injections into head and neck cancers resulted in immunomodulatory changes, with an increase in Th1 cells and activation of NK and B cells . Based on the results with i.t. or intralesional administration of IL-12, there appears to be a considerably more positive profile of therapeutic effects and considerably less SEAs than has been observed after systemic i.v. or s.c. administration.
Perhaps because the results from recent studies showed greater promise, there has been renewed interest in reevaluating IL-12 in the clinic. In 2007, the National Cancer Institute held an immunotherapy agent workshop , during which a group of renowned scientists discussed data on promising cancer immunotherapy agents, defined as “agents with known substantial immunologic or physiologic activity that have not been tested or have been inadequately tested in cancer patients,” to develop a rank ordered list of 20 reagents selected from 124 reagents considered. IL-12 was ranked third in this list for an adjuvant therapy .
Rationale for Increased Emphasis for Intratumoral Administration of Immunomodulators
The tumor microenvironment contains a complex milieu of immunologic cells and factors, with some appearing to have antitumor effects and others having immunosuppressive or immunoinhibitory effects. There is a substantial body of evidence indicating that a large proportion of tumors have a considerable number of infiltrating lymphocytes, including both specific subsets of T cells and NK cells, and patients with such infiltrates have been reported to generally have a better prognosis, including a lower risk for metastatic spread . However, despite encouraging evidence, such tumors may continue to grow progressively and metastasize. Recent studies have shown that within the tumor microenvironment, there are several mechanisms for suppressing or blocking antitumor immune responses. For example, T cells in tumors often have increased expression of inhibitory molecules such as CTLA-4 and PD-1 on T cells, and some of these T cells fall into the category of T regulatory cells (Tregs). In either case, such T cells are likely to have low or absent antitumor reactivity. In addition, there is frequently a substantial accumulation of myeloid-derived suppressor cells and tumor-associated macrophages and also high levels of suppressive cytokines such as IL-10 and transforming growth factor-β . Recently, altering the balance of immune suppression within the tumor microenvironment has gained increasing interest for enhancing antitumor immunity by i.t. injection of cytokines or other immunomodulators to alter the tumor microenvironment toward effective processing of tumor antigens by APCs, and also to augment Th1 and CTL functions, so that there can be augmented antitumor reactivity in the injected tumor and then such effector cells also distribute to other parts of the body and attack metastatic lesions . As an additional likely benefit from i.t. administration of cytokines such as IL-12, the levels of the cytokine in the injected tumor would be considerably higher at that site, with lower or negligible levels in the circulation. This should reduce the toxicity associated with systemic delivery of IL-12 or other cytokines. Methods for i.t. administration include, but are not limited to, the encapsulation of IL-12 protein into various types of microparticles . Gene therapy methods such as transfection of fibroblasts with plasmid DNA and plasmid DNA encoding IL-12 by gene gun delivery , intradermally , or directly into a tumor have also been used and may potentially provide a safer treatment option compared to systemic administration. Additional methods have included the use of novel liposome-based carriers and electroporation using plasmid DNA directly into tumor lesions . Note that traditionally, plasmid expression vectors as well as viral vectors with IL-12 have utilized transgenes with strong, constitutively activating promoters such as the cytomegalovirus promoters or elongation factor 1α. However, when constitutive promoters are utilized in vector constructs, the levels of IL-12 expression or their duration of expression cannot be controlled. Cell-based vectors for expression of IL-12 have been previously utilized with engineered fibroblasts but recently evolved to include antigen-specific T cells transduced by retrovirus or lentivirus vectors carrying the IL-12 gene, for use in T cell adoptive transfer therapies , or mesenchymal stem cells transduced for constitutive expression of IL-12 . All of these cell-based systems resulted in a reduction in tumor growth and immunomodulatory effects in murine tumor models.
An alternative approach to electroporation of plasmids or cell-based vectors has been the administration of viral-based vectors that allow for highly efficient levels of transduction. A wide variety of viral vectors have been utilized for cancer immunotherapy, such as the vaccinia/fowl pox virus , herpes simplex virus 1 , lentivirus , and Semliki Forest virus , but adenoviral vectors (Ads) have been the main viral vector used to date for transgene delivery, especially in the clinic. Regardless of the type of viral vector used for IL-12 transgene delivery in vivo , the expression of IL-12 resulted in generation of systemic antitumor immunity and therapeutic efficacy. A particular advantage of utilizing Ad is its ability to efficiently transduce both dividing and nondividing cells and the vector remains episomal, with transient expression obtained. The methods and scalable manufacturing of recombinant Ad for use in murine models and for use in patients in the clinic are easily achievable with robust and cost-effective processes, with the capability to obtain viral titers of 10 13 vector particles/ml with little or no detectable replication-competent virus in the purified product.
Dendritic Cells and Adenovirus-Transduced Dendritic Cells for Cancer Immunotherapy
DCs are powerful APCs derived from bone marrow that are capable of stimulating innate immunity and eliciting specific, adaptive T cell immune responses to various antigens, including self and tumor antigens . Various subtypes of DCs have been identified in murine and humans, ranging from the conventional myeloid DCs to plasmacytoid DCs as well as tissue-specific subtypes that can also have opposing functions . The main focus of our discussion in this chapter is on conventional myeloid DCs, derived from either macrophages or monocytes. DCs are extremely efficient in taking up antigens and processing the antigens into peptides that bind to MHC class II or class I molecules, which then are able to prime CD4 + and CD8 + T cells, respectively. DCs also have the capability for cross-presentation of exogenous antigens onto MHC class I molecules on CD8 + T cells . Increased expression of costimulatory molecules (CD80, CD86, and CD40) as part of cellular maturation also increases the polarization of T cells to Th1 and CTL, with capabilities to induce a robust T cell response. In addition to easily obtaining monocytes from blood or macrophages from bone marrow and differentiating them into DCs by culture with GM-CSF and IL-4, there are various ex vivo methods for polarizing DCs to a DC1 subset, which efficiently processes tumor antigens, secretes IL-12 and other immunomodulatory cytokines, and thereby promotes strong antitumor T cell immunity, including development of CTLs . Thus, these DCs have been attractive candidates for cancer immunotherapy. Despite the powerful immune-stimulating capabilities that DCs may possess, DCs from tumor-bearing animals as well as from patients with cancer are often found to be polarized as DC2s or to be otherwise hyporesponsive through various mechanisms . In addition, other myeloid cells within the tumor microenvironment, such as the M2 subset of macrophages, can have a greater degree of suppressive activity that further promotes tumor growth . However, there has been demonstration of the ability to shift DCs from tumor-bearers to express IL-12 and thereby promote such DCs toward a DC1-like phenotype and consequently shift CD4 + T cells toward a Th1 CD4 + T cell response and enhance the generation of CTLs .
Intratumoral injection of transduced DCs with an Ad vector that constitutively expressed IL-12 was evaluated in a murine model of CMS4 and MethA sarcomas and was found to be therapeutic, with generation of broadly reactive, tumor-specific CD8 + T cell repertoire and consequent significant tumor growth reduction . In this study, the combination of DCs expressing both IL-12 and IL-18 induced a significantly greater therapeutic response in comparison to the utilization of DCs expressing only a single cytokine . In a follow-up study using a hepatic CMS4 tumor model system, i.t. administration via intrahepatic delivery of murine Ad-IL-12-transduced DCs demonstrated reduced liver weight, as a measurement of tumor weight, and, more important, the generation of antitumor immunity involving CD4 + and CD8 + T cells and NK cells . Furthermore, when the DC IL-12-treated mice were challenged with another injection of tumor cells, they rejected CMS4 tumor cells but not Colon26 cells . One important feature in this study was the utilization of BM-derived DCs from tumor-bearing mice, which had impaired functions (i.e., allostimulation of T cells and cytokine production) compared to non-tumor-bearing mice; however, Ad-IL-12 transduction of the DCs from tumor-bearers restored the desired functionality. This finding is encouraging for analogous studies using autologous DCs for treatment of cancer patients. After in vitro co-cultures of peripheral blood mononuclear cells from melanoma patients with Ad-IL-12 (with or without the combination of Ad-IL-18), the transduced DCs induced a Th1-type response against MAGE-A6 peptide/proteins . However, in these studies, the DCs transduced by the Ad-IL-12 vector had a wide range of IL-12p70 constitutive production of IL-12, from 20 to more than 200 ng/ml during a 48-hr culture period .
Regulated Gene Expression: RheoSwitch Therapeutic System
Note that most of the studies described previously, whether employing IL-12 plasmid expression vectors, viral vectors with IL-12, or transduction with the IL-12 gene into various types of cells (e.g., fibroblasts , T cells , or mesenchymal stem cells ), utilized strong, constitutive activating promoters such as the cytomegalovirus promoters or elongation factor 1α for high levels of transgene expression. However, a major limitation to this approach is that IL-12 expression cannot be controlled in terms of the levels or duration of expression. Controlled gene transcription of cytokines, in which the transgene is placed under the control of a transcriptional switch in the promoter region of the gene and transgene expression can only be induced with a specific drug, is expected to confer an opportunity for reducing systemic toxicity. Discontinuation of administration of the drug would turn off the switch, resulting in silencing of gene expression and thereby reducing or eliminating toxic effects. An ideal transcriptional switch system would also provide a dose-dependent level of transgene expression, based on the level of activator ligand drug that is administered. Drug-inducible switch systems that have been previously utilized in in vitro and in vivo settings include (1) the tetracycline inducible (Tet)-off and Tet-on systems using doxycycline, (2) rapamycin-induced FKB12- and FRB-based systems, (3) RU486-inducible progesterone receptor-based systems, (4) tamoxifen-induced estrogen receptor-based systems, (5) the streptogramin-induced Pip repressor-based system, (6) macrolide antibiotic inducible MphR(A) protein-based systems, and (7) the ecdysone-inducible ecdysone receptor (EcR)-based system. Many of these reported systems lack at least one of the desirable characteristics mentioned previously or have not yet been sufficiently tested, especially at the clinical level, to confirm the validation of the control elements for proof of platform. Recently, molecular switches for gene expression that are based on cell differentiation or activation have also been used, including the utilization of the nuclear factor expressed in activated T cells (NFAT) promoter for conditional expression of IL-12 in activated T cells and the use of tissue-specific promoters, such as liver-specific promoters for restricted expression in the liver that can be used in conjunction with an inducible promoter system . Another approach is to constitutively express an inactive form of the gene of interest, such as caspase-9, fused with the FK506 binding protein, which would only become functional in the presence of an inert dimerizing drug .
The RheoSwitch Therapeutic System® (RTS®) is the only control switch expression platform that possesses all of the advantages and has already been validated clinically in a clinical trial involving IL-12 (discussed later). Utilization of RTS® switch with Intrexon’s Better DNA™ technology, on the UltraVector® platform, provides a novel gene-regulated system that acts as a molecular switch for strict control of inducible expression of a transgene, as outlined in Figure 25.1A . RTS® gene switch consists of three basic components: (1) an inducible promoter for the transgene of interest; (2) constitutive production of a transcription factor and coactivation partner; and (3) a novel RheoSwitch® activator ligand, termed INXN-1001, which is a diacylhydrazine small-molecule ligand that is administered orally . The transcription factor and coactivation partner of RTS® gene switch consist of two fusion proteins, Gal4-EcR and VP16-RXR. The Gal4-EcR fusion protein contains the DEF domains of a mutagenized ecdysone receptor (EcR) from the Spruce budworm ( Choristoneura fumiferana ) that is fused with the DNA binding domain of the yeast Gal4 transcription factor. The second fusion protein, VP16-RXR, consists of the EF domains of a chimeric RXR fused with the transcription activation domain of the VP16 protein of HSV1. Both of these fusion proteins are constitutively expressed, but expression of these two fusion proteins in the absence of INXN-1001 provides an “off” signal with no transgene expression. In the presence of INXN-1001, stabilization of the heterodimeric complex between the two fusion proteins forms an active transcription factor that binds to the inducible promoter and provides the “on” signal. This active transcription factor recruits transcriptional coactivators and components of the basal transcription machinery to induce expression of a target transgene placed under the control of the RTS® switch .
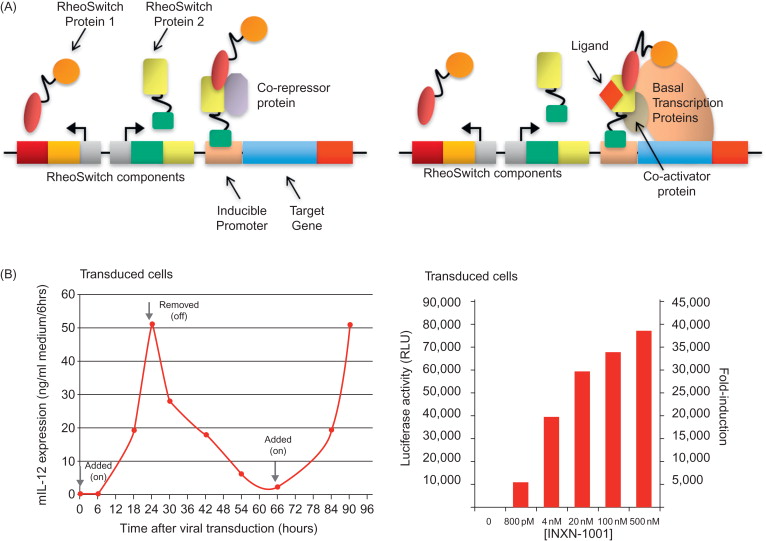
The coding sequences for both of these fusion proteins (Gal4-EcR and VP16-RXR) have been inserted in a nonreplicative Ad vector and can be expressed in host cells following Ad transduction . The Ad vector is an E1/E3 replication-defective, human serotype 5 virus that contains the gene for either murine (m)IL-12 or human (h)IL-12 under the control of RTS® switch. In the presence of INXN-1001, tumor cells transfected with an RTS-mIL-12 plasmid demonstrated regulated expression of murine IL-12. Induced expression can be detected within a few hours, with maximum expression observed within 24 hr. After INXN-1001 is removed from the culture, expression levels begin to taper off and decline to basal levels in approximately 24 hr, but the reintroduction of INXN-1001 to the cultures reinitiates the expression of IL-12 in tumor cells, demonstrating the off–on responses driven by the absence or the presence of the activator ligand ( Figure 25.1B ). In addition, the amount of transgene expression can be further regulated by the doses of INXN-1001, as demonstrated with luciferase activity in luciferase-transduced cells ( Figure 25.1B ). The utilization of RTS® switch potentially allows for regulation of transgene expression of not only IL-12 but also other genes of interest to improve safety and efficacy.
Preclinical Studies
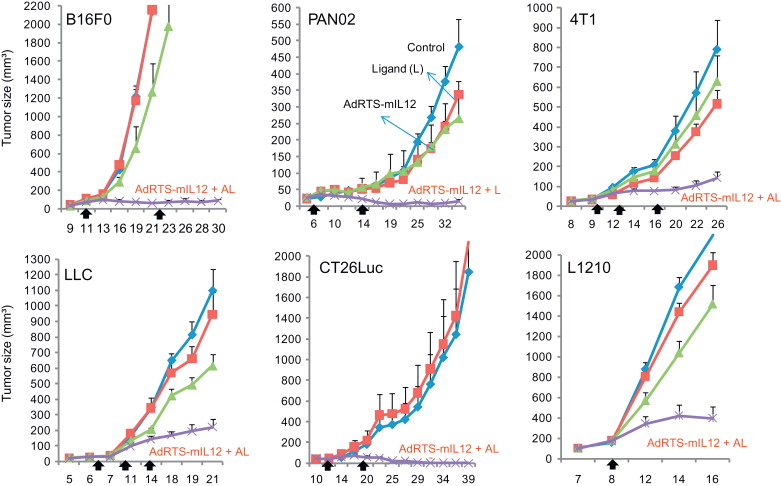
The first in vivo proof-of-concept report demonstrating the in vivo therapeutic potential involved the transduction of syngeneic BM-DCs transduced with Ad-RTS-mIL-12 and then i.t. administration of 1×10 6 DC-RTS-mIL-12 cells into an established B16F0 melanoma tumor . In mice, on day 7, when B16F0 tumors were palpable, treatment with DC-RTS-mIL-12 and with INXN-1001 by oral gavage initiated concomitantly and then continued daily for 4 more days induced expression of IL-12 in the injected tumor and provided the greatest therapeutic effect (i.e., complete tumor regression in 100% of mice). A similar degree of tumor growth inhibition was observed using DCs transduced with an Ad vector containing a constitutively expressing mIL-12 gene. In contrast, tumor-bearing mice treated with DC-RTS-mIL-12 only (i.e., with no INXN-1001 administered) or with INXN-1001 only had continued tumor growth that was similar to that of untreated controls. Delay in inducing IL-12 expression, by initiating the daily administration of INXN-1001 at 48 hr after i.t. administration, also induced significant tumor growth reduction; however, reducing the duration of INXN-1001 administration and thus the duration of IL-12 expression reduced the impact on tumor growth . The optimally effective treatment schedule for INXN-1001 was to initiate administration within 24 hr after DC-RTS-IL-12 IT injection and then to continue daily administration for a minimum of 5 days. Based on a previous report , the induction of IL-12 expression by INXN-1001 also increased the survival of the DCs in vivo . With the induction of IL-12 expression, the transduced DCs were detected by fluorescence microscopy in the draining lymph node as well as in the tumor 72 hr after i.t. injection. The increased frequency of transduced cells in the draining lymph node and tumor also corresponded with the generation of B16 tumor-specific CD8 + T cells expressing high levels of IFN-γ, suggesting that cross-priming of tumor antigens occurs and elicits a systemic antitumor immune response . Indeed, all mice treated with DC-RTS-mIL-12 remained tumor-free and failed to grow B16 tumors when rechallenged at a different site 45 days after the initial tumor injection. This resistance to tumor challenge was specific as well as durable because when the DC-RTS-mIL-12 immunized group of mice was rechallenged with the MC38 colon carcinoma cell line, tumors grew at approximately the same rate as in unimmunized mice. In a separate study, mice treated with DC-RTS-mIL-12 plus INXN-1001 in a murine firbrosarcoma (MethA) model or renal cell carcinoma (RENCA) model also demonstrated dramatic tumor growth reduction, with substantial infiltration of CD4 + and CD8 + T cells into the tumor and production of IFN-γ by the T cells . These studies demonstrate that this form of treatment, by not restricting the generated antitumor immune response to a specific tumor antigen and therefore allowing for induction of T cell immunity to a wide variety of antigens in the injected tumor, is likely to generate antitumor immunity and therapeutic efficacy in heterogeneous tumors of a particular type and can also be effective for individuals bearing different types of tumor.
Thus, DC-RTS-IL-12 vaccines are likely to represent a personalized approach, requiring the use of syngeneic or autologous DCs. To provide an alternative to the time-consuming and costly process of ex vivo differentiation and expansion of DCs followed by transduction with Ad-RTS-IL-12, the direct injection of Ad-RTS-IL-12 into a tumor lesion was hypothesized to transduce tumor cells and/or APCs in the tumor and lead to a similar induction of specific T cell antitumor immunity and therapeutic efficacy. A study by Gambotto et al. provided support for this concept by demonstrating that i.t. injection of an Ad vector containing a transgene for constitutive expression of IL-12, in day 7 established subcutaneous MCA205 fibrosarcomas, led to significant tumor growth reduction and survival, with generation of effector CTLs and enhanced infiltration of CD4 + and CD8 + T cells in the tumor . Other groups have also observed similar effects after i.t. delivery of Ad-IL-12 vectors with a constitutive promoter . To explore this approach with the Ad-RTS-mIL-12 vector, a range of doses of the viral vector particles were injected into subcutaneous established B16F0 tumors, together with oral dosing with a fixed concentration of INXN-1001. The results indicated a 73% tumor growth reduction relative to control-treated mice, using 1×10 8 viral particles, whereas greater than 90% tumor growth reduction, and tumor regression, was obtained when 1×10 9 to 5×10 10 viral particles were used . A similarly strong therapeutic effect was seen when i.t. injection of 1×10 10 Ad-RTS-IL-12 particles in combination with oral INXN-1001 were used to treat various established s.c. tumor models, which included syngeneic murine tumor cells lines 4T1 (breast), RENCA (renal cell carcinoma), PAN02 (pancreatic), L1210 (lymphocytic leukemia), LLC (Lewis lung carcinoma), and CT26 (colon carcinoma), as shown in Figure 25.2 . The levels of tumor growth inhibition were significantly greater in the Ad-RTS-mIL-12 with oral INXN-1001-treated group compared to the control-treated groups .
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Ethics in Translational Gene Transfer Research
Clinical Trials Using LV-P140K-MGMT for Gliomas
Ethics in Translational Gene Transfer Research
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


