Keywords
Cancer, vaccines, poxviruses, TRICOM, immunotherapy, intratumoral vaccination, immunostimulatory molecules, costimulatory molecules
In this chapter, we review the use of recombinant poxviral vectors containing transgenes for costimulatory molecules and transgenes for tumor-associated antigens (TAAs) to enhance the immunogenicity of tumors. This is accomplished either by infecting tumor cells ex vivo with these recombinant (r) vectors or by direct intratumoral (i.t.) vaccination. Also discussed are the use of these recombinant vectors as “conventional” therapeutic vaccines when administered subcutaneously (s.c.) or in combined approaches employing concomitant s.c. and i.t. vaccination. We review studies principally carried out in our program at the National Cancer Institute and with our collaborators. Both preclinical and clinical studies are reported as well as proposed approaches for future clinical trials.
The Use of Recombinant Poxviral Vectors
We hypothesized that the use of vectors that inherently enhance immunity would act to also enhance the immunogenicity of an inserted tumor-associated transgene. Vaccinia virus is one of the most immunogenic viruses known. It is the agent used in the worldwide eradication of smallpox and has been administered to more than 1 billion humans. Vaccinia virus and other members of the poxvirus family are attractive vectors for numerous reasons, as outlined in Table 22.1 .
| Large amounts of foreign DNA can be inserted under the control of individual poxvirus promoters. |
| Recombinants are stable. |
| Replication is accurate with efficient host translational processing of inserted genes. |
| They have a wide cell range and host range. |
| They can efficiently infect antigen-presenting cells such as dendritic cells and tumor cells. |
In addition to recombinant vaccinia, we have also employed two additional recombinant poxviral vectors in our studies. Modified vaccinia Ankara (MVA) is an attenuated form of vaccinia virus. It is replication defective and is currently considered the “safe smallpox vaccine.” However, it retains much of the immunogenicity of intact vaccinia virus. Avipox viruses, fowlpox and canarypox (ALVAC), are replication incompetent in mammalian cells. They are thus considered to be quite safe. Although antibody responses to avipox vectors are induced in mammalian species post-vaccination, they are non-neutralizing due to the fact that late viral proteins (i.e., viral coat proteins) are not translated in mammalian cells.
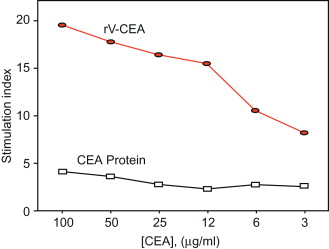
In one of the initial experiments, we evaluated the ability of a recombinant vaccinia to enhance immunogenicity of a transgene. We used carcinoembryonic antigen (CEA) as a prototype TAA. CEA is overexpressed in a wide range of human carcinomas and is also expressed in human fetal tissue and gastrointestinal (GI) tissue. The CEA-transgenic (Tg) mouse, expressing the human CEA gene in both fetal and GI tissue, was employed. As seen in Figure 22.1 , vaccination with CEA protein in adjuvant was not able to break tolerance to this “self” antigen, whereas rV-CEA induced appreciable CEA-specific T cell immunity.
T Cell Costimulation
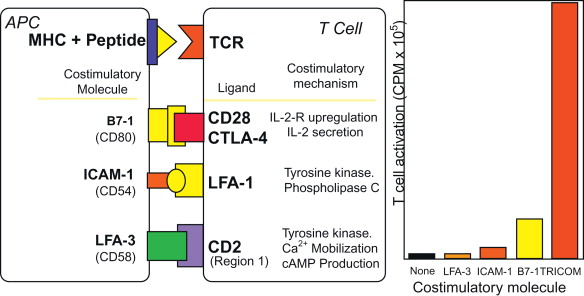
The generation of a robust host response to a weak “self-antigen” such as tumor-associated antigens requires at least two signals. The first signal is the engagement of the T cell receptor (TCR) via the peptide major histocompatibility complex (MHC), and a second signal involves the engagement of a costimulatory molecule on the antigen-presenting cell (APC) with its ligand on the T cell. B7.1 (CD80) is one of the most studied costimulatory molecules in its interaction with its CD28 ligand on T cells. An rV-B7.1 vector was first constructed and shown to faithfully express B7.1 transgenes. We then constructed recombinant vaccinia vectors containing transgenes for B7.2, ICAM-1, LFA-3, and CD70, among others, and demonstrated that each of these could enhance T cell responses to a test antigen .
The Use of Vectors Containing Multiple Costimulatory Molecules
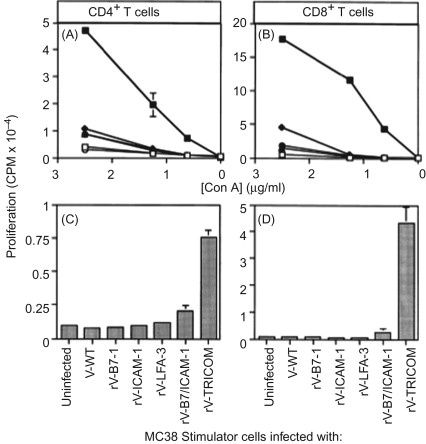
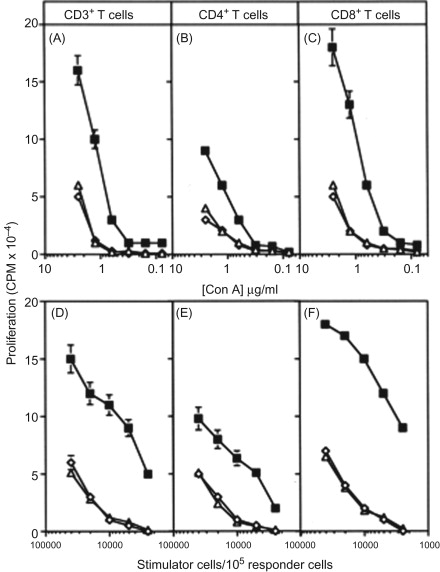
We then engineered two recombinant poxvirus vectors (a replication-defective avipox (fowlpox; rF) and a replication-competent vaccinia (rV)) to express a triad of murine costimulatory molecules (B7.1, ICAM-1, and LFA-3; designated TRICOM). We compared the ability of these TRICOM vectors to that of vectors containing one costimulatory molecule—that is, with rV- and rF-B7.1, or ICAM-1, or LFA-3 alone, or in combination (i.e., rV- or rF-B7.1/ICAM-1)—to activate both CD4 + and CD8 + T cell responses . Both rV- and rF-TRICOM vectors clearly outperformed the other recombinants in numerous T cell activation assays ( Figure 22.2 ). The level of activation by TRICOM was clearly synergistic in that T cell activation was greater than that achieved by adding the levels of activation seen by rV- or rF-B7.1, plus rV- or rF-ICAM-1, plus rV- or rF-LFA-3 ( Figure 22.3 ). Levels of T cell activation by TRICOM were most dramatic under conditions of low levels of antigen or low stimulator cell:T cell ratios. These studies were the first to demonstrate the ability of vectors to introduce three costimulatory molecules into a cell and to efficiently activate both CD4 and CD8 T cell populations to far greater levels than that achieved by any one or two costimulatory molecules.
Tricom Vector Infection of Dendritic Cells
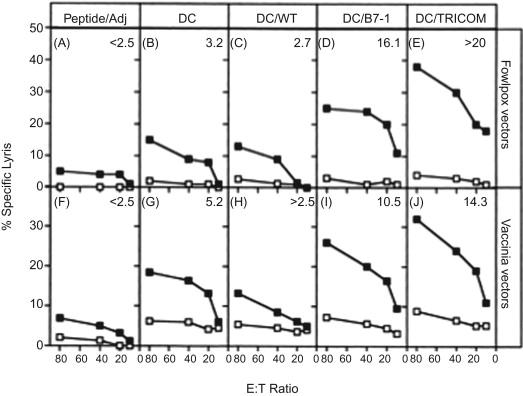
Dendritic cells (DCs) constitutively express several costimulatory molecules and are believed to be the most potent APCs. A study was designed to determine whether DCs infected with these TRICOM vectors would have an enhanced capacity to stimulate T cell responses . Murine DCs (of both intermediate maturity and full maturity) were infected with rF-TRICOM (mu) or rV-TRICOM (mu) and were used in vitro to stimulate naive T cells with the use of a pharmacologic agent as signal 1, to stimulate T cells in allospecific mixed lymphocyte cultures, and to stimulate CD8 + T cells specific for a peptide from the ovalbumin (OVA) protein. In addition, DCs infected with TRICOM vectors were pulsed with OVA peptide and used to vaccinate mice to examine T cell responses in vivo . Dendritic cells infected with either rF-TRICOM or rV-TRICOM were found to greatly enhance naive T cell activation ( p <0.001), allogeneic responses of T cells ( p <0.001), and peptide-specific T cell stimulation in vitro ( p <0.001). Peptide-pulsed DCs infected with rF-TRICOM or rV-TRICOM induced cytotoxic T lymphocyte (CTL) activity in vivo to a markedly greater extent than peptide-pulsed DCs ( p =0.001) ( Figure 22.4 ).
Tricom Infection of Non-DC APCs
Although DCs are arguably the most potent APCs, limitations clearly exist in their use due to the level of effort and cost for their generation. We conducted studies to demonstrate that a generic APC population, such as murine splenocytes, can be made markedly more efficient as APCs by infection with either rF-TRICOM or rV-TRICOM vectors ( Figure 22.5 ) . Infection of murine splenocytes with either rV- or rF-TRICOM (mu) vector led to significant improvement of APC capabilities in terms of (1) enhancement of mixed lymphocyte reactions, (2) a reduction in the amount of signal 1 needed to activate naive T cells, and (3) a reduction in the amount of APCs required to activate T cells using a constant amount of signal 1. TRICOM-enhanced T cell activation was shown to correspond to increases in type 1 cytokines and a reduced level of apoptosis compared with T cells activated with uninfected or control vector-infected splenocytes. In vitro and in vivo experiments compared the use of DCs with that of TRICOM-infected splenocytes. Infection of splenocytes with TRICOM vectors markedly enhanced their ability to activate T cells to levels approaching that of DCs. These studies thus demonstrated for the first time that an abundant and accessible population of APCs obtainable without lengthy culture or the use of costly exogenous cytokines (in contrast to that of DCs) can be made more potent as APCs with the use of vectors that express the triad of costimulatory molecules constituting TRICOM.
Whole Tumor Cell Vaccines
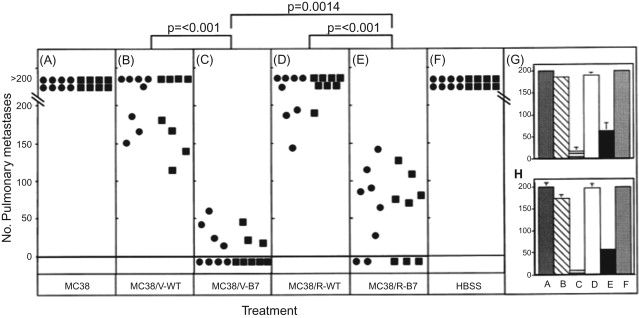
We have conducted studies to compare for the first time the use of a retroviral vector versus a vaccinia to enhance the immunogenicity of whole tumor cell vaccines . The tumor cell used was the weakly or nonimmunogenic MC38 murine colon adenocarcinoma. Infection of murine carcinoma cells with low multiplicity of infection of wild-type vaccinia virus (V-WT) had no therapeutic benefit upon subsequent tumor transplantation. In contrast, inoculation of rV-B7.1-infected tumor cells into immunocompetent animals resulted in no tumor growth .
Using live whole tumor cells as vaccine, cells transduced via recombinant retrovirus (MC38/R-B7) or recombinant vaccinia (MC38/V-B7) equally induced protection against subsequent challenge by native MC38 cells 14 days later. Upon rechallenge with native MC38 cells 40 days later, however, the MC38/R-B7 vaccine was shown to be less effective than the MC38/V-B7 vaccine. Similar results were obtained when the tumor cells were irradiated prior to administration. When studies were conducted in which X-irradiated tumor cell vaccines were given to mice bearing experimental lung metastases, the MC38/V-B7 vaccine was shown to be significantly more effective than the MC38/R-B7 vaccine ( Figure 22.6 ). Additional studies were carried out in mice that had received vaccinia virus previously to render them vaccinia immune. Again, the X-irradiated MC38/V-B7 vaccine was statistically ( p =0.024) more effective than the MC38/R-B7 vaccine in the elimination of metastases. These studies thus demonstrated for the first time that a whole tumor cell vaccine (either live or X-irradiated) containing a vaccinia costimulatory molecule transgene is at least as effective, and sometimes more effective, at inducing antitumor effects as the same vaccine using a retrovirus to express the transgene.
Enhancing the Immunogenicity of Chronic Lymphocytic Leukemia Cells: Potential for a Tumor Cell Vaccine
Chronic lymphocytic leukemia (CLL) is a disease of CD5 + B lymphocytes (designated as CLL cells) that are inefficient APCs. Their poor ability to present antigens to the T cells, largely due to an inadequate costimulatory capacity, is manifested as a failure to stimulate proliferation of both allogeneic and autologous T cells. A recombinant MVA, which is a highly attenuated, replication-impaired virus variant, was successfully used to infect and deliver the simultaneous expression of the three human costimulatory molecules in TRICOM on the surface of the CLL cells . Proliferation of allogeneic and autologous T cells was observed when MVA-TRICOM-infected CLL cells were used as stimulators in proliferation assays ( Table 22.2 ). Cytotoxic T lymphocytes, generated in vitro by stimulation of autologous T cells with MVA-TRICOM-infected CLL cells, showed cytotoxicity against unmodified/uninfected CLL cells. These findings suggested that the use of CLL cells infected ex vivo with MVA-TRICOM or direct injection of MVA-TRICOM in patients with CLL has potential for the immunotherapy of CLL. We also demonstrated for the first time that T cells from CLL patients can acquire multiple costimulatory molecules from autologous CLL cells and can then act as APCs .
| Patient No./Stimulator Cells | Autologous Stimulation Index |
|---|---|
| Pt. 2 | |
| Uninfected | 1.0 |
| MVA-WT | 0.5 |
| MVA-TRICOM | 34.8 |
| Pt. 8 | |
| Uninfected | 1.3 |
| MVA-WT | 1.6 |
| MVA-TRICOM | 30.6 |
| Pt. 16 | |
| Uninfected | 1.7 |
| MVA-WT | 1.3 |
| MVA-TRICOM | 27.2 |
a CD3 + T cells from patients with CLL were stimulated with uninfected, MVA-WT-, or MVA-TRICOM-infected CLL cells at a ratio of stimulator to effector cells equal to 1:2.5. Proliferation was measured as 3 H-thymidine incorporation on day 5 of culture. Stimulation index was calculated as cpm (T cells+CLL cells)/cpm (T cells alone).
In another study, we compared MVA encoding CD40L or TRICOM for their ability to enhance the immunogenicity of CLL cells. CLL cells from some patients showed differential responses to each vector in terms of induction of autologous T cell responses . These studies provide the rationale for the use of CLL cells modified ex vivo with prespecified recombinant MVA vectors as a whole tumor cell vaccine for immunotherapy in CLL patients.
The Generation of Tricom-Based Vaccines: Preclinical Studies
A series of preclinical murine studies was conducted with recombinant vaccinia viruses containing transgenes for T cell costimulatory molecules and transgenes for TAAs. These included recombinant vaccinias expressing LFA-3, CD70, ICAM-1, 41BBL, and OX-40L . Each was shown to enhance specific T cell responses. Through a series of in vitro and in vivo preclinical studies, we determined that the combined use of the three costimulatory molecules in TRICOM (B7.1, ICAM-1, and LFA-3) acted synergistically to enhance antigen-specific T cell responses. Each of these molecules binds to a different ligand on T cells and has been shown to signal through a different pathway ( Figure 22.3 ). It is interesting to note that attempts to add a fourth costimulatory molecule to TRICOM resulted in only a minimally enhanced immunogenicity and in one case a reduction in immune responses to a transgene.
Many of the “proof-of-concept” preclinical studies with poxviral vectors have been conducted employing recombinant poxviruses expressing the human CEA transgene and its ability to induce immunity in a mouse transgenic for human CEA under the control of the endogenous CEA promoter. As mentioned previously, as in humans, the CEA-Tg mouse expresses high levels of CEA in fetal tissues and GI epithelium. Vaccination with rV-CEA, but not CEA protein in adjuvant, could induce CEA-specific T cell responses. Studies then demonstrated that the admixing of rV-CEA with rV-B7.1 resulted in enhanced CEA-specific T cell responses and antitumor immunity compared to either rV-CEA or rV-B7.1 vector alone . One of the concerns regarding the use of costimulatory molecules in vaccines is the potential for the induction of autoimmunity. A toxicology study was conducted and it was determined that multiple vaccinations of rV-B7.1 induced no toxicity in a murine model.
The Generation of High-Avidity CTLS
High-avidity CTLs are most effective at clearing viruses and cancer cells. Therefore, understanding the mechanisms involved in induction of high-avidity CTLs is critical for effective vaccines. Signals from MHC class I (signal 1) and costimulatory molecules (signal 2) were adjusted by varying antigen (Ag) dose and by the use of the TRICOM (mu) vectors . A strong signal 1 resulted in an increased frequency of CD8 + CTLs. However, a strong signal 2 was necessary for the induction of high-avidity CD8 + CTLs that killed target cells more efficiently, and signal 2 played a more crucial role in the presence of a weak signal 1. Only CTLs induced with strong signal 2 killed tumor cells endogenously expressing low levels of antigen. Signal 2 contributed to the induction of high-avidity CD8 + CTLs in both primary and secondary responses. Thus, although signal 2 has been known to increase the quantity of CTL response, this study was the first to show that it also improves the quality of CTL response.
Diversified Prime and Boost Regimens
The property of intense immunogenicity of the replication-competent vaccinia, however, also serves to limit the number of inoculations. Preclinical and clinical studies have shown that in a vaccinia-immune host (i.e., a patient who had received the smallpox vaccine), recombinant vaccinia can be given only once, or at most twice, to enhance immunogenicity of the tumor antigen transgene. After that, host immunity to vaccinia limited the host’s ability to mount immunity to the transgene. Recombinant avian poxviruses (avipox) such as fowlpox (rF-) or canarypox (ALVAC) have been used to potentiate immunity to transgenes following rV- priming. The use of a diversified immunization schema in CEA-Tg mice using rV-CEA as a prime followed by multiple boosting vaccinations with avipox CEA was shown to be far superior to the use of either one alone in eliciting a CEA-specific T cell response . The superiority of this diversified schema has been demonstrated using TRICOM-based vaccines in both preclinical models and clinical trials.
We have used different vaccines and vaccine strategies in an attempt to develop optimal antitumor immunity in the stringent CEA-Tg animal model. These studies demonstrated that (1) continued boosting with vaccine is required to maintain CEA-specific T cell responses, and following vaccination with rV-CEA-TRICOM, boosting with rF-CEA-TRICOM is superior to boosting with rF-CEA; and (2) a diversified vaccination protocol consisting of primary vaccination with rV-CEA-TRICOM followed by boosting with rF-CEA-TRICOM is more efficacious than homogeneous vaccination with either agent alone in the induction of both CEA-specific T cell responses and antitumor activity ( Figures 22.7 and 22.8 ) .
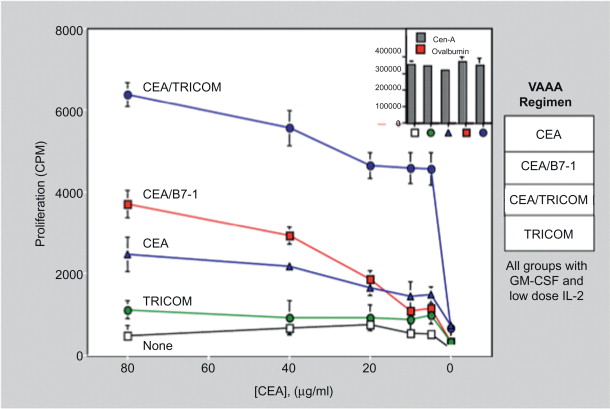
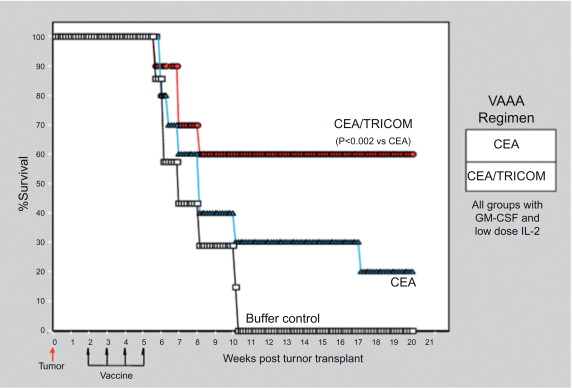
Tricom-Based Vaccines: Clinical Studies
Three of the most widely studied human TAAs are CEA, mucin-1 (MUC-1), and prostate-specific antigen (PSA). CEA is overexpressed in a wide range of human carcinomas, including GI, breast, lung, pancreatic, medullary thyroid, ovarian, and prostate. MUC-1 is a tumor-associated mucin that is overexpressed and hyperglycosylated in all human carcinomas as well as in acute myeloid leukemia and multiple myeloma. The elegant studies of Kufe and colleagues as well as others have demonstrated that the C-terminus of MUC-1 functions as an oncogene . PSA is overexpressed in the vast majority of prostate carcinomas as well as in normal prostatic epithelium.
We conducted the first trial in humans of the CEA-TRICOM vaccines (also including an enhancer agonist epitope within the CEA gene) . Fifty-eight patients with advanced CEA-expressing cancers were accrued to eight cohorts that involved vaccinations with the following: multiple vaccinations with rF-CEA(6D)-TRICOM; primary vaccination with rV-CEA(6D)-TRICOM followed by rF-CEA(6D)-TRICOM booster vaccinations; and rV-CEA(6D)-TRICOM and then rF-CEA(6D)-TRICOM, plus granulocyte–macrophage colony-stimulating factor (GM-CSF) with vaccines, or with divided doses of vaccine with GM-CSF. Vaccines were administered every 28 days for six doses and then once every 3 months. Reverting to treatments every 28 days was allowed if patients progressed on the 3-month schedule.
No significant toxicity was observed. Twenty-three patients (40%) had stable disease for at least 4 months, with 14 of these patients having prolonged stable disease (>6 months) ( Figure 22.9 ). Eleven patients had decreasing or stable serum CEA, and 1 patient had a pathologic complete response. Enhanced CEA-specific T cell responses were observed in the majority of patients tested. We thus demonstrated that the CEA-TRICOM vaccines are safe and can generate significant CEA-specific immune responses, and they may have clinical benefit in some patients with advanced cancer.
Related posts:
 Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
Modified Oncolytic Herpesviruses for Gene Therapy of Cancer
 Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
Genetically Engineered (T Cell Receptor) T Cells for Adoptive Therapy
 Clinical Trials Using LV-P140K-MGMT for Gliomas
Clinical Trials Using LV-P140K-MGMT for Gliomas
 Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
Therapeutic Efficacy and Systemic Antitumor T Cell Immunity Induced by RheoSwitch-Regulated IL-12 Expression after Intratumoral Injection of Adenovirus Vector or Vector-Transduced Dendritic Cells
 Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
Viral Insertion Site Detection and Analysis in Cancer Gene Therapy
 Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Selectively Replicating Oncolytic Adenoviruses Combined with Chemotherapy, Radiotherapy, or Molecular Targeted Therapy for Treatment of Human Cancers
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


