Fig. 11.1
Schema for testing agents through the Pediatric Pre-clinical Testing Program (PPTP). Adapted from reference [68]
Standard Cytotoxic Agents
Responses to standard cytotoxic agents (vincristine, cyclophosphamide, actinomycin D, cisplatinum, and topotecan) are depicted in “heat map” format in Fig. 11.2. These agents were evaluated in up to 51 PDX models, with 184 drug/tumor studies available for analysis. Actinomycin D was evaluated in only the sarcoma and kidney tumor panels (Wilms tumors and rhabdoid tumors), whereas the other agents were screened more broadly. Each of the agents showed quite robust activity against tumor types where they are used as standard-of-care therapy. For example, vincristine, an anti-mitotic tubulin-binding agent, is highly active in ALL models and has activity in pPDX models derived from medulloblastoma, rhabdomyosarcoma, and Wilms tumors. Cyclophosphamide also has broad-spectrum activity, whereas cisplatinum has activity in several solid tumor panels, but is poorly active against the ALL models, consistent with its lack of clinical utility in childhood ALL [72]. Topotecan also showed solid tumor and ALL activity consistent with clinical data. The overall objective response rate (≥Partial Response) was 39.1% (72/184 tumor/drug studies) and serves as a benchmark for evaluating nonstandard cytotoxic agents.
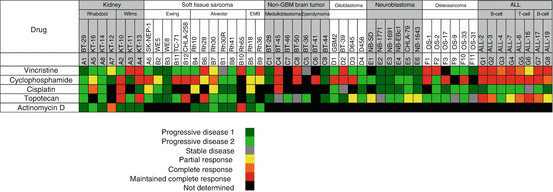
Fig. 11.2
Heat map representation of anti-tumor activity of standard of care cytotoxic agents. Tumor panels are arrayed horizontally at the top, and specific PDX and cell-derived xenograft models are shown for each tumor panel. Drugs are listed in the left column. Tumor response classification is shown below
Novel Cytotoxic Agents
The activity of 19 novel cytotoxic agents with different mechanisms of action is shown in Fig. 11.3. There were 481 tumor/drug evaluations, and the overall response rate was 25.4%. Most of these agents have not been evaluated in Phase II clinical trials in children, so it is not possible to determine the validity of the pPDX results. However, the proteasome inhibitor, bortezomib, showed activity against only ALL models, and this is consistent with single-agent clinical activity [73]. Two agents are useful for illustrating the value and limitations of pre-clinical xenograft models. The pre-prodrug, PR-104 is activated under hypoxic conditions to 5-hydroxylamine (PR-104H) and amine (PR-104M) to produce DNA interstrand cross-links [74, 75]. This agent was tested at the maximum-tolerated-dose level as, at that time, human pharmacokinetic data were not yet available. The drug demonstrated dramatic broad-spectrum activity (kidney tumors, sarcoma, and ALL). However, it transpired that at the MTD in mice, drug exposures were four- to five-fold above exposures achieved in (adult) patients at the recommended Phase II clinical trial dose level. Subsequent dose response data showed that at dose levels in mice corresponding to human systemic exposures, significant anti-tumor activity was observed only in T-cell ALL models. Of interest, T-cell ALL has very high levels of aldo-keto reductase 1C3 (AKR1C3) that also activates PR-104 [76], suggesting that tumor-selective anabolism in T-cell ALL is responsible for sensitivity to this agent [76].
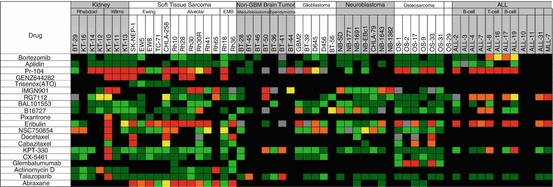
Fig. 11.3
Heat map representation of anti-tumor activity of novel cytotoxic agents. Tumor panels are arrayed horizontally at the top, and specific PDX and cell-derived xenograft models are shown for each tumor panel. Drugs are listed in the left column. Tumor response classification is as shown in Fig. 11.1
The second agent is the tubulin binder eribulin and can be used to illustrate the positive and negative values of pre-clinical models in developing anti-mitotic agents. This drug has a mode of binding to tubulin that is distinct from vincristine, which for pediatric cancer is the gold standard. In the screen, eribulin showed marked activity in numerous tumor panels at dose levels below the MTD. Notably, eribulin was active in five of six Ewing sarcoma xenograft models, whereas vincristine was not active (see Fig. 11.3). Pharmacokinetic modeling suggests that the systemic exposure to drug in mice is relevant to clinical exposure [77]. However although eribulin caused regression in several osteosarcoma PDX models, it has not shown activity in a Phase II clinical trial in osteosarcoma, suggesting the models overpredict activity. Indeed, vincristine showed activity against osteosarcoma models (Fig. 11.2) but has not been efficacious when added to other combinations in clinical trials. While the models may overpredict activity of anti-mitotic agents, it is of note that the rate of regression in the osteosarcoma models were far slower than in soft tissue models where tumors completely regressed with 1–2 weeks of starting treatment. However, the osteosarcoma results raise the question of whether the PDX models overpredict for the activity of anti-mitotic agents. As shown in Fig. 11.3, cabazitaxel and docetaxel caused tumor regressions but at a dose/schedule that gave exposures per 21-day cycle well above systemic exposures tolerated in patients [78], consistent with the lack of activity of taxane drugs in pediatric cancer. In contrast, another anti-mitotic tubulin binder, BAL101553, showed virtually no anti-tumor activity against either solid tumor or ALL models [79]. The PPTP also tested nanoparticle albumin-bound paclitaxel (nab-paclitaxel; Abraxane) in sarcoma models (Fig. 11.3). Systemic exposures in mice were about 50% higher in mice than patients: (nab-paclitaxel (50 mg/kg) was 36.54 μM*Hr in mice. In contrast human exposure was 23.80 μM*Hr in patients receiving 260 mg/m [80]. It is unclear whether plasma exposures are important for this agent as it appears to be concentrated in tumor tissue. Hence tumor drug levels in pPDX models relative to patient tumors may be more relevant. However, that nab-paclitaxel had significant anti-sarcoma activity at drug exposures close to that in patients, in contrast to docetaxel and cabazitaxel, suggests this agent may have activity in Ewing sarcoma and rhabdomyosarcoma.
Signaling Inhibitors
Twenty-four signaling inhibitors have been evaluated as single agents through the PPTP (Fig. 11.4). These included small-molecule inhibitors and antibodies. Overall there were 1272 drug/tumor data sets that included up to 53 xenograft models. There were 61 objective responses (4.1%), significantly less than observed with the novel cytotoxic agents. Of note, three agents that target the process of mitosis were the most active. GSK923295A, an inhibitor of the centromere kinesin motor protein CENP-E, the Aurora kinase A inhibitor MLN8237 (alisertib), and the polo-like kinase inhibitor BI6727 (volasertib) each showed considerable activity when tested at the MTD. However, for each drug, it is estimated that plasma exposures were fivefold greater than were achieved in patients. For alisertib, the dose-response curve was steep, and at 25% of the MTD, very few pPDX models, other than ALL, were responsive [81]. GSK923295A was also active only at the mouse MTD [82], and volasertib was not tested at lower dose levels as this agent had limited activity at doses resulting in drug exposures tenfold greater than in adult patients. If responses induced by these three agents are eliminated as being “false positives,” the objective response rate (ORR) for signaling inhibitors decreases to 26/1152 drug/tumor tests (2.25%). This raises the question of whether the PDX/xenograft models overpredict for cytotoxic drugs, but underpredict for signaling inhibitors. Clinical experience shows that for several signaling inhibitors tumor regressions are usually associated with a particular molecular characteristic. Examples are the activity of trastuzumab in HER2-amplified breast carcinoma [83], imatinib in BCR/Abl-activated chronic myelogenous leukemia [84], erlotinib in ERBB1-mutated NSCLC [85], crizotinib and other ALK inhibitors in EML4-ALK-translocated NSCLC [86], or BRAF/MEK inhibitors in BRAF-mutated melanoma [87]. For pediatric cancers, particularly solid tumors, the frequency of activating mutations that would predispose to drug sensitivity seems to be quite low. One exception is low-grade glioma where activation of BRAF occurs through tandem duplication of the BRAF:KIAA1549 locus [88] or point mutation (usually V600E) in most tumors. In the PPTP screen, the MEK inhibitor selumetinib (AZD6244, Fig. 11.4) had very little activity [89], whereas in secondary screening against two astrocytoma models, it was highly active against the astrocytoma model with a BRAF-activating mutation (BRAFV600E), but had no activity against the model with wild-type BRAF (Fig. 11.5). Of interest, recently, it was reported that at relapse, there was a higher incidence of Ras pathway activation, potentially sensitizing tumors to MEK inhibitors [64]. In the PPTP in vitro screen, RD cells (NRas mutant) and NB-EBc1 (KRas mutant) were significantly more sensitive to selumetinib. However, NB-EBc1 xenografts were not responsive and neither was the Rh36 embryonal rhabdomyosarcoma PDX that has an HRas mutation. Other results that also suggest that the models accurately predict for activity of signaling inhibitors are the sensitivity to sunitinib of ALL-2, a line with FLT3 mutation (Y572S) [90], sensitivity of NB-1643 with an ALK (R1725Q) mutation to the ALK inhibitor crizotinib [91], and sensitivity to dasatinib in a Ph+ALL model.
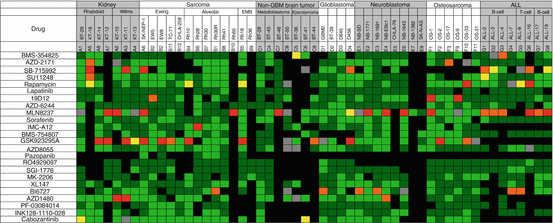
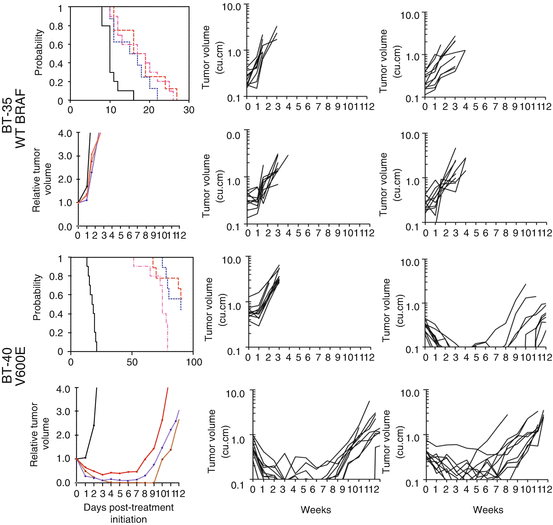
Fig. 11.4
Heat map representation of anti-tumor activity of signal transduction inhibitors. Tumor panels are arrayed horizontally at the top, and specific PDX and cell-derived xenograft models are shown for each tumor panel. Drugs are listed in the left column. Tumor response classification is as shown in Fig. 11.1
Fig. 11.5
AZD6244 activity against pilocytic astrocytoma xenografts. Kaplan-Meier curves for EFS, median relative tumor volume graphs, and individual tumor volume graphs are shown for (a): BT-35 and (b) BT-40. Kaplan-Meier curves: controls, black solid line; 100 mg/kg BID × 5/SID × 2 for 6 consecutive weeks, broken red line; 75 mg/kg BID × 5/SID × 2 for 6 consecutive weeks, blue broken line; 100 mg/kg SID for 6 consecutive weeks, broken pink line. Relative tumor volume curves: controls, black solid line; 100 mg/kg BID × 5/SID × 2 for 6 consecutive weeks, solid brown line; 75 mg/kg BID × 5/SID × 2 for 6 consecutive weeks, solid blue line; 100 mg/kg SID for 6 consecutive weeks, solid red line. For individual growth curve plots: upper left panel, control; upper right panel, AZD6244 100 mg/kg BID × 5/SID × 2 for 6 consecutive weeks; lower left panel, AZD6244 75 mg/kg BID × 5/SID × 2 for 6 consecutive weeks; lower right panel, 100 mg/kg SID for 6 consecutive weeks. Adapted from reference [89]
One aspect of translating pre-clinical results is that schedules of administration used in mice may not be possible, or reproduced, in clinical protocols. An example is alisertib that induced objective responses in six of six ALL models at its MTD, in three models at 0.5 MTD, and in two of three at 0.25 MTD using twice daily dosing, 5 days per week for 3 consecutive weeks [81]. In the trial in childhood ALL (NCT01154816), the schedule was amended to once daily for 7 days, due to increased toxicity in pediatric patients. The amended schedule, once daily for 7 days, achieved one response in 33 patients and hence was not considered active. A retrospective study, in two ALL PDX models sensitive to the initial schedule of administration, showed neither models were sensitive to the revised schedule, daily for 7 days [92].
Combination Testing
Constraints of clinical testing in pediatric cancer include the paucity of patients eligible for experimental drugs, and testing of these agents is usually conducted in a refractory or relapse setting. Consequently, identification of active agents seems to be relatively slow. When we developed the topoisomerase I poisons, topotecan and irinotecan, there was clear evidence for activity in Phase I clinical trials, and these agents were further tested in “upfront window” trials against drug naive patients with advanced disease [29, 30, 93]. In general, a new agent will be added to existing regimens. For example, we evaluated topotecan and irinotecan combination with standard agents, cyclophosphamide and vincristine, used routinely in the treatment of childhood solid tumors [14, 20]. The combination of vincristine and irinotecan (VI) is now incorporated into several protocols for treatment of rhabdomyosarcoma and has recently been shown as highly active in anaplastic Wilms tumor [36]. The mechanism for synergy between camptothecin derivatives such as topotecan and irinotecan and vincristine remains to be elucidated, but this combination seems to be active in several childhood cancers. Similarly, the combination of irinotecan with temozolomide had greater than additive activity in PDX models [37, 94] and has been introduced into several protocols for solid tumors at relapse with or without vincristine [40–42, 95].
The design of drug combination studies is critical, and demonstrating therapeutic synergy requires multiple dose levels of each agent alone or in combination. Many studies, especially those reporting modulation or reversal of multi-drug resistance (MDR), used flawed experimental designs (reviewed in [96]). Essentially, in all published studies (at that time), the MDR modulator + cytotoxic drug was compared to the cytotoxic drug at the same dose used in the combination—despite the combination with the modulator necessitating the reduction of the dose of cytotoxic agent from its optimal dose (maximum tolerated dose level, MTD). For the PPTP, a method was developed to identify “therapeutic enhancement” [23]. Simply stated the combination had to have greater effect than either single agent used at its MTD.
Relatively few combination studies were conducted through the PPTP. Notable were studies with cediranib (AZD2171), a potent small-molecule inhibitor of vascular endothelial growth factor (VEGF) receptors. The cediranib-cyclophosphamide combination was inferior to single-agent cyclophosphamide for both models studied and was significantly inferior for one of the models [97]. In contrast, rapamycin enhanced the therapeutic activity of both cyclophosphamide (Fig. 11.6) and vincristine against several PDX models [23]. For treatment of sarcoma at relapse, the Children’s Oncology Group (COG) has adopted an approach where a novel agent is added to a “backbone” therapy for relapsed patients. Using a two-arm design, it is possible to compare the activity of two experimental agents simultaneously (drug A + backbone therapy versus drug B + backbone therapy). The NCT01222715 trial used a backbone of cyclophosphamide + vinorelbine, adding the rapalog temsirolimus to one arm and bevacizumab to the other arm for relapsed rhabdomyosarcoma. The trial was stopped when it became clear that the temsirolimus arm was significantly superior to that containing bevacizumab. These results seem to parallel the pre-clinical predictions, that is, rapamycins enhance the activity of Vinca alkaloids and cyclophosphamide, whereas an inhibitor of VEGF signaling was antagonistic to cyclophosphamide. However, without a control arm (cyclophosphamide + vinorelbine only), it is not possible to determine whether temsirolimus enhanced the activity of backbone therapy or whether bevacizumab antagonized backbone therapy.
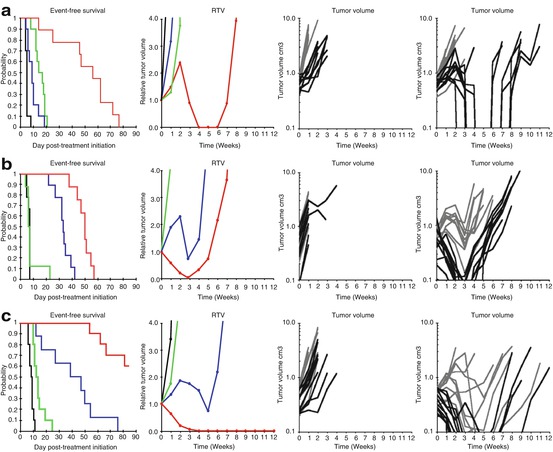
Fig. 11.6
Rapamycin enhances the therapeutic activity of cyclophosphamide. Tumor-bearing mice were treated with cyclophosphamide at the MTD (150 mg/kg daily dosing 7 days per week for 6 consecutive weeks), rapamycin (5 mg/kg d × 5 for 6 consecutive weeks), or the combination of cyclophosphamide and rapamycin. Tumor diameters were measured weekly. Panel 1 shows the Kaplan-Meier curves for EFS: control (black), rapamycin (green), cyclophosphamide (blue), or rapamycin + cyclophosphamide (red). Panel 2 shows median relative tumor volumes: control (black), rapamycin (green), cyclophosphamide (blue), or rapamycin + cyclophosphamide (red). Individual tumor-growth curves are shown in panel 3, control (light gray) and rapamycin (dark lines), and panel 4, cyclophosphamide (light gray) and cyclophosphamide + rapamycin (dark lines). (a) D456 glioblastoma; (b) KT-14 rhabdoid tumor of kidney; (c) Rh30 rhabdomyosarcoma
Rapamycin also appears to enhance the activity of irinotecan in several PDX models, although, again, the mechanism seems obscure. Indeed, based on the cell cycle-dependent killing by topoisomerase I poisons, one would have anticipated some antagonism as rapamycin causes an increase in G1 fraction, whereas irinotecan (or more precisely the active metabolite SN-38) kills cells only during DNA replication (S phase). In yeast rapamycin potentiates the toxicity of methylmethane sulfonate (MMS)-induced DNA damage, possibly through suppressing damage-induced subunits of ribonucleotide reductase and preventing error-prone trans-lesion bypass repair [98]. Of interest, the combination induced greater kill than did MMS alone but, importantly, suppressed MMS-induced mutations. Thus, because rapalogs enhance the efficacy of DNA-damaging agents such as cyclophosphamide and irinotecan and show therapeutic enhancement in some PDX models, it is proposed to test temsirolimus in a backbone of vincristine-irinotecan in a future COG rhabdomyosarcoma trial.
Interest in developing inhibitors of poly-ADP ribose polymerase (PARP) was largely stimulated following the observation of a synthetic lethal interaction in cell lines lacking homologous recombination through loss of BRCA function [99, 100]. Several PARP inhibitors have demonstrated single-agent activity in patients where BRCA1 or BRCA2 homozygous mutations are tumor specific [101, 102]. Ewing sarcoma cell lines were also reported to be hypersensitive to PARP inhibitors [103, 104], possibly as a result of an interaction between PARP1 and EWS-FLI1. Brenner et al. [104] also reported a dramatic synergy between temozolomide and olaparib in a xenograft model of Ewing sarcoma, although olaparib as a single agent had little activity.
The PARP1/2 inhibitor, talazoparib, was evaluated by PPTP as a single agent, in 44 xenograft models representing childhood solid tumors and ALLs. The result of this testing showed very modest activity among all models. Tumor regressions were observed only in a medulloblastoma and a Wilms tumor PDX [105]. The PPTP data seem to be reflective of clinical activity since continuous high-dose olaparib had no activity in a Phase II clinical trial for Ewing sarcoma patients [106]. There are several pre-clinical studies that provided evidence for combination of PARP inhibitors with DNA damaging agents as chemotherapy with promising activity in xenograft tumor models. This strategy was also supported by the emerging evidence that PARP1 inhibitors are cytotoxic due to PARP trapping initiated by PARP1 binding to DNA with single-strand breaks [107].
Talazoparib showed the highest efficiency at trapping PARP-DNA complexes among all other known PARP inhibitors [108]. Temozolomide contributes to the process by producing N3 and N7 methyl adducts of adenine and guanine, respectively. N7-methylguanine and N3-methyladenine adducts, the most abundant and nonlethal lesions, are repaired by BER, but the process requires PARP to recognize and bind the repair intermediate 5′-deoxyribose phosphate. Thus, PARP inhibitors turn the nonlethal N7-methylguanine and N3-methyladenine into cytotoxic lesions by trapping PARP at 5′-deoxyribose phosphate [109, 110]. Recruitment of additional PARP molecules critical for BER is impaired by inhibition of PARP’s catalytic activity; single-stranded breaks become double-stranded breaks, which require homologous recombination for repair. Subsequently, the ability to potentiate temozolomide toxicity aligns with PARP-trapping capacity of PARP inhibitors [107, 108]. Overall, combination of temozolomide with such a potent PARP-trapping inhibitor as talazoparib is a more rational approach than combination of temozolomide with a PARP inhibitor that primarily acts by catalytic inhibition.
In our in vitro studies, the low-level damage to DNA induced by temozolomide was potentiated up to 85-fold through inhibition of PARP by talazoparib in the PPTP studies [111]. The potentiation was seen in Ewing sarcoma cell lines as well as ALL cell lines. However, despite significant modulation in vitro, the only objective regressions induced by the talazoparib/temozolomide combination were in Ewing sarcoma xenografts, with no activity in ALL. Two dose levels of temozolomide were used in PPTP testing in vivo, 30 mg/kg and 12 mg/kg, both administered for 5 consecutive days. Talazoparib was administered at a lower dose with “high”-dose temozolomide. Overall, talazoparib was equally effective when used in a low-dose temozolomide compared to a high-dose temozolomide regimen (Fig. 11.7), which is consistent with the emerging understanding of the role of PARP trapping for talazoparib. Both combinations demonstrated significant activity against five out of ten Ewing sarcoma xenograft models and only modest activity against rhabdomyosarcoma, glioblastoma, and Wilms tumor models, which may suggest that resistance mechanisms in the unresponsive lines exist de novo. In the TC-71 Ewing sarcoma model, essentially complete acquired resistance to the combination of talazoparib and temozolomide developed within five transplant generations. Our preliminary data support the mechanism of PARP1/2 trapping on DNA, rather than inhibition of catalytic activity, being responsible for cytotoxicity in Ewing sarcoma models (RTK unpublished).
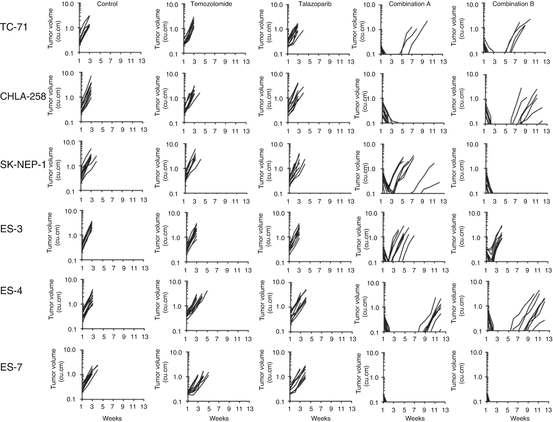
Fig. 11.7
Responses of ‘sensitive’ Ewing sarcoma xenografts to single-agent therapy or the combination of talazoparib plus temozolomide. Anti-tumor activity of temozolomide (30 mg/kg D×5); talazoparib (0.25 mg/kg BID D×5); or in combination against ‘sensitive’ Ewing sarcoma models. Combination A (temozolomide 30 mg/kg D×5; talazoparib 0.1 mg/kg BID D×5); Combination B (temozolomide 12 mg/kg D×5; talazoparib 0.25 mg/kg BID D×5). Graphs show growth of individual tumors in SCID mice (from reference 111)
The PPTP results were used to develop the talazoparib/temozolomide Phase I/II clinical trial through the COG (NCT02116777). As in mice, combination of temozolomide with talazoparib necessitated temozolomide dose reduction to overcome high toxicity of the combination. Early data from the trial indicated thrombocytopenia is limiting when talazoparib (600 μg/m2) is combined with temozolomide (30 mg/m2), approximately 15% of the MTD for single-agent temozolomide. This is a similar dose reduction from the MTD for temozolomide, as in mice, when combined with talazoparib.
Chemoradiation Combinations
Radiation therapy (RT) remains an essential component of curative therapy for most solid tumors of childhood, yet the long-term consequences of RT, particularly for patients with CNS tumors, can be devastating. For treatment of medulloblastoma, even after reducing the RT dose to 23.4 Gy, neurocognitive outcome data revealed a significant decline of about 1.7 points per year in intellectual functioning over 5 years and an IQ drop of 10–20 points in younger children [112]. The long-term consequences of chemoradiation treatment strategies include neuroendocrine-cognitive deficits, visual deficits, seizure disorders, motor disturbances, vasculopathy, and secondary tumors [113, 114]. Necrotizing leukoencephalopathy [115], a diffuse white matter injury, was first described in children receiving intra-thecal methotrexate [115] with subsequent combined RT and chemotherapy. It has also been observed in adult patients with leukemia [116] and various malignancies that had metastasized to the CNS [117]. The intent for combining RT with molecularly-targeted agents is to increase the cure rate and to reduce the dose of RT through radiosensitization. The PPTP studies did not include RT or RT combinations. However, in limited studies, following up on PPTP findings, we were able to demonstrate synergistic activity of RT when combined with the MDM2 inhibitor RG7388 [118] in a rhabdomyosarcoma pPDX and with selumetinib (MEK inhibitor) in a heterotopic model of BRAF mutant astrocytoma [119]. Selumetinib enhanced the RT activity by a factor of two- to threefold in this model. The importance of the latter study is that it may be possible to maintain tumor control but reduce the RT dose and hence reduce the long-term CNS toxicities described above. However, it will be critical to demonstrate that inhibition of MEK does not potentiate RT damage to normal brain.
Molecular Subgroup Representation
One of the limitations of the PPTP is that while we evaluated drugs against more than 50 pPDX models, there were limited models representing any one pediatric cancer. For example, the PPTP used eight ALL models, but usually only two models represented ependymoma, medulloblastoma, or Wilms tumor. Clearly this does not represent the clinical heterogeneity of these pediatric tumors. Initially tumor models were compared to clinical samples to determine whether expression profiles were representative of clinical disease [15]. Subsequently, models were characterized by expression profiling and single-nucleotide polymorphisms [120]. Overall, the results suggested that the PDX and xenograft models were similar to the clinical disease from which each was derived. However, to be truly representative of the clinical heterogeneity, further model development is required. Thus, within resource constraints, how can the molecular heterogeneity of pediatric cancer in a pre-clinical screen be accomplished? One approach has been initiated by Novartis, where a large number of melanoma PDX models were developed. Screening used an individual mouse as a “patient.” In this case, 30 melanoma PDX models, each derived from different patients, were grown in a single mouse, and the objective response (≥50% volume regression) rates (ORR) were assessed for different agents [121]. In part, this would be equivalent in design to a clinical Phase II trial where ORR is determined. The advantage of this approach is that far greater molecular heterogeneity can be encompassed and perhaps gives a more precise estimate of the likely clinical response rate, assuming that the pharmacology of a drug in mice is similar to that in patients. This experimental design identifies drugs that induce a high frequency of tumor regressions, but because no control arm is used, slowing of tumor progression is not identified. The other advantage over the traditional experimental design (more mice/group but fewer tumor lines) is that the potential to identify “exceptional responders” is increased and hence the potential to identify underlying characteristics that would allow clinical stratification in subsequent trials.
In a retrospective analysis of 67 agents tested by the PPTP, we determined whether a single mouse, chosen randomly from each group of a study, predicted the median response for groups of mice with tumor [122]. The individual tumor response from one randomly-chosen mouse was compared to the group median response using criteria developed in the PPTP. A total of 2134 comparisons were made. The single-tumor response accurately predicted the group median response in 1604 comparisons (75.16%), and the mean-tumor response correct-prediction rate for 1000 single-mouse random samples was 78.09%. Models had a range for correct prediction (60%–87.5%). Allowing for mis-prediction of ± one response category, the overall mean-correct single-mouse prediction rate was 95.28%. Further, from the single-mouse data, the predicted overall objective response rates for group data were correct for 66 of 67 drug studies. No differences between pPDX and cell line-derived xenografts were found. Assuming that large treatment effects are targeted, this alternate experimental design has similar predictive value as traditional approaches, allowing for far greater numbers of models to be used that more fully encompass the heterogeneity of disease types. One would anticipate that a “hit” in the single-mouse screen would be confirmed using a traditional experimental design (using 8–10 mice per tumor line).
Development of Drug Resistance
One of the limitations in pediatric oncology is the inability to access patient tumor at relapse, in most instances because of ethical considerations. However, this severely limits the ability to identify drug resistance mechanisms in a cohort of uniformly treated patients receiving protocol therapy. It also limits the ability to develop PDX models from patients at relapse that would more appropriately represent a typical Phase II clinical trial patient population. Such efforts are underway for several adult cancers in so-called co-clinical trials [123, 124]. We have used PDX models to develop drug resistance to vincristine, melphalan, and selumetinib, respectively, in tumor models initially highly sensitive to the individual agent [24, 25, 125]. The approach was to treat tumor-bearing mice with drug at a dose and schedule causing CR, then stopping treatment and allowing tumor to regrow. The tumor with most rapid regrowth was transplanted and the process repeated. This approach was used to select resistance to the talazoparib-temozolomide combination discussed above and for development of resistance to irinotecan (CPT-11) as illustrated in Fig. 11.8. However, the approach is somewhat limited if a traditional experimental design is used (i.e., one would develop resistance to agents in one or two models). On the other hand, coupled to the “single-mouse” design discussed above, it may be valuable for determining predominant mechanism(s) of action to single agents and combinations in a larger number of PDX models (each derived from a different patient tumor), thus simulating potential mechanisms of clinical drug resistance rather than that developed in vitro under conditions of increasing drug selection pressure.
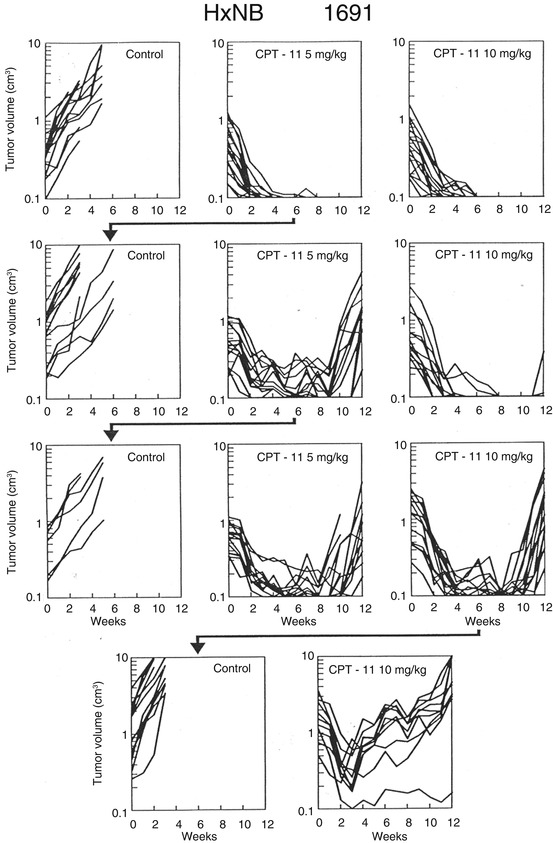
Fig. 11.8
 Cervical Cancer PDOX Models
The Revival of Patient-Derived Xenograft Mouse Models of Cancer: Way Back to the Future
Use of Patient-Derived Orthotopic Xenografts (PDOX) to Evaluate Transformative Cancer Therapeutics
Why Patient-Derived Mouse Models Need to Be Orthotopic
Cervical Cancer PDOX Models
The Revival of Patient-Derived Xenograft Mouse Models of Cancer: Way Back to the Future
Use of Patient-Derived Orthotopic Xenografts (PDOX) to Evaluate Transformative Cancer Therapeutics
Why Patient-Derived Mouse Models Need to Be Orthotopic
 3D Cell Culture Models
3D Cell Culture Models
 Development of Orthotopic and Spontaneous Metastatic Human Tumor Xenograft Models for Experimental Therapeutics
Development of Orthotopic and Spontaneous Metastatic Human Tumor Xenograft Models for Experimental Therapeutics




Development of resistance to irinotecan (CPT-11) in NB-1691 xenografts. The schema shows response of parental NB-1691 neuroblastoma to irinotecan (CPT-11). Irinotecan was administered parenterally (IP) 5 days per week for 2 consecutive weeks. Tumor from mice treated at 5 mg/kg was transplanted (arrow). Tumor in the subsequent passage was less sensitive to treatment, and again a regrowing tumor from the 5 mg/kg group was transplanted and retested for drug sensitivity. In this passage, a tumor that regrew at the 10 mg/kg dose level was transplanted and its sensitivity tested in the subsequent passage, where it was relatively resistant to this dose of irinotecan
Related posts:
Cervical Cancer PDOX Models
The Revival of Patient-Derived Xenograft Mouse Models of Cancer: Way Back to the Future
Use of Patient-Derived Orthotopic Xenografts (PDOX) to Evaluate Transformative Cancer Therapeutics
Why Patient-Derived Mouse Models Need to Be Orthotopic
3D Cell Culture Models
Development of Orthotopic and Spontaneous Metastatic Human Tumor Xenograft Models for Experimental Therapeutics
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
