Fig. 11.1
Available drugs in clinical trials to block the Ubiquitin-Proteasome System. All members of the ubiquitin family are generated through a high molecular weight precursor that has to be cleaved by specific proteases to generate the mature form of each modifier. The covalent attachment of ubiquitin is mediated by a thiol-ester cascade of reactions involving at least three enzymes: an activating enzyme (AE) or E1; a conjugating enzyme (CE) or E2; and a ligase (LE) or E3. Ubiquitin-like proteins such as SUMO or NEDD8 have their own set of enzymes. Demodifying enzymes such as DUBs, SUSPs or NEDP1 participate in a proofreading mechanism. Multiple chain types can be formed and the composition of the chains is essential to drive distinct functions, including proteolysis by the 26S proteasome
11.1.3 Proteasomes
The 26S proteasome is a multi-subunit complex formed by a barrel-shaped proteolytic core, the 20S core particle, and one or two regulatory 19S particles flanking the ends of the core to regulate the entry of proteins targeted for degradation (Fig. 11.1) (Bedford et al. 2010). The 20S core particles consist of four stacked heptameric ring structures that are themselves composed of two different types of subunits. The two outer rings are formed by seven alpha subunits (α1–α7) that allow the interaction with the 19S particle, while the inner rings are composed of seven beta subunits (β1–β7), three of which are responsible for the catalytic activity. The catalytic mechanism of these subunits is the same because they use the hydroxyl group of the N-terminal threonine of a mature β-subunit to perform the nucleophilic attack on the carbonyl carbon of a peptide bond (Borissenko and Groll 2007; Kisselev and Goldberg 2001; Kisselev et al. 2012; Voges et al. 1999). Each subunit reacts with specific substrates: β1 subunit possesses peptidyl-glutamyl peptide-hydrolyzing (PHGH) activity that cleaves after acid residues, β2 has trypsin-like activity that cleaves after basic residues and β5 subunit shows chymotrypsin-like activity that cleaves after hydrophobic residues. Site-directed mutagenesis studies of these catalytic residues in yeast showed that the most drastic changes occurred when β5 subunit is inactivated (Heinemeyer et al. 1997).
The 19S is divided into two sub-particles, the base and the lid. The lid contains at least nine non-ATPase polypeptide chains (Rpn3, 5–9, 11, 12 and 15) that remove the polyubiquitin chains from the protein-substrates, while the base consists of four non-ATPase (Rpn1, Rpn2, and Rpn13) and six ATPase subunits (Rpt1–Rpt6) that interact directly with α-rings. These ATPases control the opening of the 20S gate that is normally locked until an unfolded substrate is recognized. Only the unfolding step involves ATP hydrolysis (Peth et al. 2010).
Apart from the proteasome, an immuno-proteasome exists in immune cells where the 11S subunit, also known as PA28 or REG, replaces the 19S subunit. The 11S is dominantly expressed in hematopoietic cells in response to pro-inflammatory signals such as interferon gamma or cytokines, and is involved in antigen processing for subsequent presentation to the MHC-I on the cell surface, allowing the initiation of the immune cell response (Rock et al. 1994; Tanaka and Kasahara 1998). Furthermore, the immune-proteasome facilitates the clearance of protein aggregates to prevent cell death produced by IFN-induced oxidative stress, (Seifert et al. 2010). All proteasome inhibitors currently used in clinical trials block the enzymatic activities of the proteasome. However, the next generation of proteasome inhibitors will block other mechanism such as gate opening or regulatory subunits.
11.2 Targeting Proteasomes
The first proteasome inhibitors made available were synthesized to specifically block the proteasome’s active sites and to understand its enzymatic mechanism. Peptide aldehydes such as MG-132 were used to develop analogues with enhanced potency, selectivity and stability. Surprisingly, several studies showed that proteasome inhibitors induced rapid and selective apoptosis in different cancer derived cell lines, leading to the idea that proteasome inhibitors could be drug candidates. This idea was confirmed by the fact that the proteasome inhibitor Bortezomib regressed tumour size of xenograft tumours and also decreased metastasis and blocked angiogenesis in patients with hematologic malignancies. Thus, Bortezomib was the first proteasome inhibitor approved by the Food and drug Association of the United States (FDA). Since its approval in 2003, it became the frontline treatment for MM, it has been accepted as second line treatment of MCL and it has also been included in hundreds of on-going clinical trials. In addition to Bortezomib, Carfilzomib, another proteasome inhibitor, has been approved for relapsed/refractory MM, and other proteasome inhibitors are in clinical and preclinical trials (Table 11.1).
Table 11.1
Proteasome inhibitors in clinical trials
Drug name | Company | Molecular target | Family structure | Binding type | Adms route | Clinical status | Disease | Ref |
|---|---|---|---|---|---|---|---|---|
Bortezomib (Velcade) | Takeda | Proteasome β5 > β1> β2 | Boronate | Reversible | IV SC IP | Launched | Cancer: Multiple myeloma, non-Hodgkin’s lymphoma | Adams et al. (1998) McBride and Ryan (2013) Frankland-Searby and Bhaumik (2012) |
Phase II | Cancer: B-cell lymphoma, Hodking’s lymphoma, leukaemia chronic lymphocytic, leukaemia acute myelogenous, small cell lung, non-small cell lung, prostate, gastrointestinal, breast, renal, liver, head and neck, ovarian, colorectal, oesophageal, pancreatic, sarcoma, bladder, glioblastoma, tyroid, osteosarcoma, mesothelioma, myelodyplastic syndrome, waldenstrom macroglobulinemia, unspecific solid tumor | |||||||
Others: Lupus, HCV, HIV, cytomegalovirus infections, thrombotic disorders | ||||||||
Phase I | Cancer: Melanoma, testicular, endometrial, metastatic, medulloblastoma | |||||||
Carfilzomib (PR171) | Onyx | Proteasome β5 >> β1, β2 | Epoxyketone | Irreversible | IV | Launched | Cancer: Multiple Myeloma | Frankland-Searby and Bhaumik (2012) McBride and Ryan (2013) Demo et al. (2007) Xolalpa et al. (2013) Pautasso et al. (2013) |
Phase II | Cancer: Non-Hodgkin’s lymphoma, leukaemia chronic lymphocytic, small cell lung, non-small cell lung, renal, unspecified solid | |||||||
Others: Cytomegalovirus infections | ||||||||
Phase I | Cancer: Hodgkin’s lymphoma, leukaemia acute lymphocytic, leukaemia acute myelogenous | |||||||
MLN9708 (Ixazomib) | Takeda | Proteasome β5 > β1 > β2 | Boronate | Reversible | IV | Phase III | Cancer: Multiple myeloma | Frankland-Searby and Bhaumik (2012) McBride and Ryan (2013) Kupperman et al. (2010) Xolalpa et al. (2013) |
Oral | Phase II | Cancer: Non-Hodgkin’s lymphoma, leukemia acute myelogenous | ||||||
Phase I | Cancer: Hodgkin’s lymphoma, leukemia acute lymphocytic, leukemia chronic lymphocytic, waldenstrom’s hypergammaglobulinaemia, non-small cell lung, colorectal, melanoma, renal, sarcoma, prostate, pancreatic, head and neck, endometrial, breast, gastric, esophageal, testicular, liver, thyroid, unspecified solid | |||||||
CEP-18770 (delanzomib) | Teva | Proteasome β5 > β1 > β2 | Boronate | Reversible | IV | Phase II | Cancer: Multiple myeloma | Xolalpa et al. (2013) Piva et al. (2008) Frankland-Searby and Bhaumik (2012) |
Oral | Phase I | Cancer: Non-Hodgkin’s lymphoma, non-small cell lung, colorectal, head and neck, ovarian, renal, prostate, unspecified solid | ||||||
ONX-0912 (oprozomib) | Onix | Proteasome β5 | Epoxyketone | Irreversible | Oral | Phase II | Cancer: multiple myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, leukemia chronic lymphocytic, leukemia chronic myelogenous, waldenstrom’s hypergammaglobulinaemia | Xolalpa et al. (2013) Frankland-Searby and Bhaumik (2012) Chauhan et al. (2010) |
Phase I | Cancer: non-small cell lung, liver, prostate melanoma, ovarian, renal, gastric, colonorectal, medulloblastoma, breast, sarcoma, unspecified solid | |||||||
NPI-0052 (marizomib) | Nereus | Proteasome β5 > β2> β1 | Β-lactone | Irreversible | IV | Phase I | Cancer: Multiple myeloma, non-Hodgkin’s lymphoma, Hodgkin’s lymphoma, leukaemia chronic lymphocytic, leukaemia acute lymphocytic, leukaemia acute myelogenous, non-small cell lung, sarcoma, pancreatic, melanoma, prostate, liver, ovarian, breast, renal, gastric, unspecified solid | Xolalpa et al. (2013) Fenical et al. (2009) McBride and Ryan (2013) Frankland-Searby and Bhaumik (2012) |
Oral | ||||||||
Syrbactins, Pono Pharma | Pono Pharma | Proteasome | Syrbactin | Irreversible | Unspecified | Preclinical | Cancer: Myeloma, unspecified | Groll et al. (2008) |
Proteasome inhibitor IkerChem | IkerChem | Proteasome | Unspecified | Unspecified | Unspecified | Preclinical | Cancer: Haematological unspecified, solid unspecified | |
Peptide epoxyketones Onyx Pharmaceuticals | Onyx Pharmaceuticals | Proteasome | Epoxyketone | Unspecified | Unspecified | Preclinical | Immunological: Inflammatory disease unspecified | |
ONX-0914 ( PR-957) | Onyx Pharmaceuticals | Inmunoproteasome β5i/LMP7 | Epoxyketone | Irreversible | Unspecified | Preclinical | Immunological: Arthritis, rheumatoid, psoriasis, inflammatory bowel disease, lupus erythematosus | Verbrugge et al. (2012) |
Cancer: haematological, unspecified | ||||||||
FV-162 | Fluorinov Pharma | Proteasome β5 | Fluorine | Irreversible | Oral | Preclinical (Phase I planned) | Cancer: Myeloma, non-Hodgkin’s lymphoma | |
Bortezomib micelle | NanoCarrier | Proteasome β5 > β1> β2 | Boronate | Reversible | Unspecified | Preclinical | Cancer: unspecified | |
Timosaponin AIII (BN-108) | Bionovo | Proteasome | Steroidal saponin | Unspecified | Oral | Preclinical (Phase I/II planned) | Cancer: breast | Chun-Nam et al. (2011) |
11.2.1 Proteasome Inhibitors in Clinical Use
The new generation of proteasome inhibitors always aims to increase potency, specificity and stability with good bioavailability and pharmacokinetics. Conventional proteasome inhibitors efficiently block at least one of the three proteasome active sites (β1, β2 and β5) either with covalent or non-covalent, reversible or irreversible binding. Most Inhibitors and drugs in clinical trials are mimetic peptides that interact directly with the active site thus blocking the nucleophilic attack of the hydroxyl group of the proteasome N-terminal threonine active sites (Kisselev and Goldberg 2001; Orlowski and Kuhn 2008). Proteasome inhibitors are often classified according to their functional group (Table 11.1):
Boronic acid peptides: Peptide aldehyde analogues were synthetized substituting the aldehyde group for boronic acid (Bortezomib) (Adams et al. 1998). Bortezomib is more potent, stable and interacts more specifically with the β5 subunit, forming tetrahedral intermediates with two extra hydrogen bonds that stabilize the covalent bond (Groll et al. 2006). Bortezomib is a reversible inhibitor administrated intravenously with very low dissociation rate that behaves as an irreversible molecule. It has a dose limiting toxicity and produces multiple side effects in patients, including pain, fatigue, gastrointestinal, cardiovascular and pulmonary disorders, neutropenia, thrombocytopenia and peripheral neuropathy. Despite its potency and effectiveness, about 60 % of the treated patients develop resistance to Bortezomib after 1 year of treatment (Mujtaba et al. 2012). The resistance mechanisms are still poorly understood but have a multifactorial basis (Xolalpa et al. 2013).
A second-generation of proteasome inhibitors, currently in clinical trials, include the boronic acid peptides MLN2238, MLN9708 and CEP–18770. Unlike Bortezomib, these inhibitors reduce neuropathy. MLN2238 and its oral analogue MLN9708, are reversible inhibitors in phase III trials with stronger chymotrypsin-like activity inhibition in vivo and a faster dissociation rate, able to penetrate inside tissues (Kupperman et al. 2010). The oral inhibitor CEP-18770 in phase I and II trials shows encouraging results for the treatment of haematologic and solid tumours (Piva et al. 2008).
Epoxyketone peptides. These peptides are the most potent and specific proteasome inhibitors known. They form a morpholine adduct with the N-terminal threonine through a covalent and an irreversible bond (Groll et al. 2000). Carfilzomib (Demo et al. 2007) and ONX–0912 (Chauhan et al. 2010) belong to this class of inhibitors. They show a high chemical stability causing a stronger inhibition of the chymotrypsin-like proteasome activity than Bortezomib (Huber and Groll 2012), and overcome the problem of resistance to Bortezomib. Due to their higher specificity, neurotoxicity is reduced (Molineaux 2012) but other side effects still persist (Fostier et al. 2012). Carfilzomib is currently used for relapsed and refractory multiple myeloma and the orally bioavailable ONX-0912 is included in phase I and II trials where it has shown an improved therapeutic window.
β– lactones : Non–peptidic inhibitors have also been developed to improve bioavailability. The compound derived from the marine microorganism Salinispora tropica, Marizomib, is a β-lactone inhibitor in phase I trials (Fenical et al. 2009). Its β-lactone group reacts with the catalytic threonines of the proteasome active site forming an acyl-ester adduct and a tetrahydrofuran ring that stabilizes the binding leading to a highly potent, selective and orally bioactive proteasome inhibitor. Contrary to epoxyketones and peptide boronic acids, Marizomib inhibits the three activities of the proteasome irreversibly, in a stronger (more than 90 %) and longer-lasting way (Miller et al. 2011). The use of marizomib results in adverse effects such as fatigue, nausea, vomiting and dyspnea.
11.2.2 Development of New Proteasome Inhibitors
A second generation of proteasome inhibitors is being designed to specifically inhibit a subunit of interest, to reduce toxicity in normal cells and increase apoptosis in tumour cells (Parlati et al. 2009). This is the case of FV–162 that shows a better inhibition of chymotrypsin-like with an improved safety profile compared to carfilzomib, and the epoxyketone ONX 0914. ONX 0914 is a potent drug orally bioavailable for the immunoproteasome and has been developed for the treatment of autoimmune disorders.
Moreover, natural products and non-covalent proteasome inhibitors are also being developed to reduce side effects. Timosaponin AIII is an example of a natural product in preclinical development whose phases I/II in breast cancer are under preparation. Withaferin A and gambonic acid are other natural inhibitors, which have been approved by the Chinese FDA for cancer clinical trials.
To avoid resistance to proteasome inhibitors, and to limit the off-target effects, the development of compounds acting in a non-competitive way that do not directly interact with the active catalytic β subunit’s site, has been considered. These compounds could potentially be used in combination with Bortezomib to improve clinical outcomes. For example, Rapamicin and PR–39 are compounds that bind to the α7 subunits, thereby producing a disruption in the interaction between 20S and 19S regulatory subunits that blocks the entry of the substrate (Gaczynska et al. 2003). 5–amino–8–hydroxyquinoline also interacts with the α-subunits inside the proteolytic chamber; while being cytotoxic for myeloma and leukemia cells in vitro, it has also been shown to decrease tumour size in xenograft tumour growth in vivo (Li et al. 2010), and overcomes resistance to Bortezomib in cultured cell lines.
11.2.3 Cellular Effects of Proteasome Inhibitors
Proteasome inhibitors are successful in cancer cells with high proliferative rates. Although all mechanisms are not fully understood, it seems that they block the degradation of proapoptotic proteins and cell cycle regulators (Fig. 11.2). Among them, the tumor suppressors retinoblastome (Rb), p53 and the cyclin-dependent kinase inhibitors (CDKIs), such as p21Cip1, p27Kip1 and p57Kip2 are degraded by the proteasome (Frankland-Searby and Bhaumik 2012; Rastogi and Mishra 2012; Vlachostergios et al. 2013). Regulators of the intrinsic apoptotic pathway are also controlled by the proteasome, including members of the Bcl-2 protein family (p53-target-genes) such as the proapoptotic proteins Bax, Bid, Noxa or Bad, and anti-apoptotic members such as Bcl-XL and Bcl-2. Bortezomib treatment increases the expression of proapoptotic members such as Bax, Smac and Noxa while anti-apoptotic proteins like Bcl-2 and IAPs are down-regulated, thus favoring apoptosis in tumor cells (Crawford and Nahta 2011).
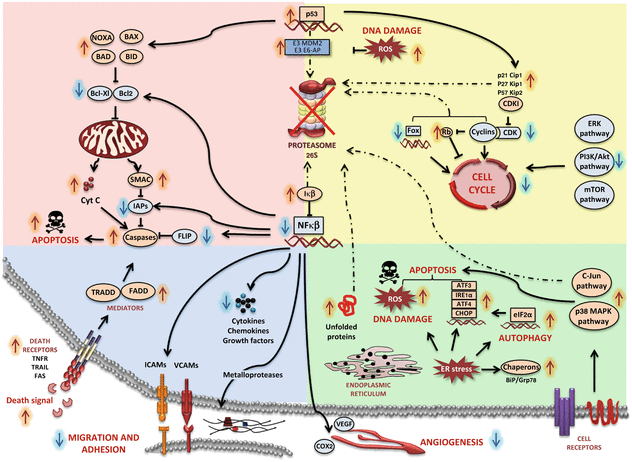
Fig. 11.2
Cellular effects of proteasome inhibition. Inhibition of the proteasome stabilizes and activates critical factors that control several apoptotic pathways in cancerous cells. Most pathways affected are involved in cell cycle control, cell proliferation, oncogenic activity, apoptosis, tumour suppressor functions and includes other multiple regulatory proteins (see main text)
The mechanism to induce apoptosis of each proteasome inhibitor seems to be different, thus some combinations result in synergies. For example, Carfilzomib and Marizomib induce apoptosis mediated by caspase 8 and 9 better than Bortezomib, which is more dependent on Bax and Bak mitochondrion-mediated cell death (Kuhn et al. 2007; Miller et al. 2007).
The inhibition of NFκB by Bortezomib seems to have an important role in this drug’s mechanism of action, although it cannot explain all the anticancer effects (Fig. 11.2). NF-κB-mediated transcription plays a crucial role in tumorigenesis by controlling among others the immune and inflammatory responses; apoptosis suppression (by inducing anti-apoptotic IAPs and BCL-XL proteins); angiogenesis promotion (by inducing vascular endothelial growth factor VEGF and COX2); favouring cell proliferation (by increasing cyclin D) and increasing cell migration (by inducing metallo-proteases) (Roccaro et al. 2006). Furthermore, constitutive expression of NF-κB has been related with resistance against radiation and chemotherapy in various types of cancer (Sunwoo et al. 2001).
Disruption of the proteasome activity also leads to the aggregation of unfolded proteins, thereby producing ER stress. An extensive protein production and secretion has been found in different types of cancer, becoming more sensitive to ER stress (Hoeller and Dikic 2009). ER stress leads to up-regulation of the endoribonuclease/kinase IRE1a and the transcription factors ATF3&4 that will increase transcriptional activation of the pro-apoptotic Noxa (Armstrong et al. 2010). Autophagy is also induced by ER stress as a resistance mechanism to escape cell death through the eIF2α induced pathway, the activation of HDAC6, the IRE1-JNK pathway, by proteasomal stabilization of ATF4, the inhibition of mTOR, and by the reduced proteasomal degradation of LC3 (Belloni et al. 2010). The activation of p38 MAPK and c-Jun N-terminal kinases has been also reported after proteasome inhibition. This leads to 14-3-3 phosphorylation and consequently to cytochrome c release. The generation of reactive oxygen species (ROS) after the treatment with proteasome inhibitors results in the expression of apoptosis-related proteins (Fan et al. 2011). ROS are responsible for DNA damage and proteasome plays a crucial role in DNA repair through the activation of p53 and other pathways.
11.2.4 Experience with Clinical Trials
Bortezomib has shown promising activity and a durable response as a single agent in relapsed or refractory MCL and MM patients (Table 11.1). Trials in Phase I demonstrated an effective plateau at 65–70 % proteasome inhibition with fair tolerance levels. In phase II, 35 % of patients with refractory MM and 46.5 % of patients with pre-treated MCL responded to Bortezomib, therefore the FDA initially approved it for relapsed/refractory MM in 2003, and subsequently for relapsed/refractory MCL in 2006 (Kane et al. 2006). Moreover, survival rates of refractory MM patients using Bortezomib in phase III trials exceeded the response rates for patients treated with the previous drug choice Dexamethasone, resulting in Bortezomib being approved as a first-line therapy in 2008 (Richardson et al. 2005). However the refractory response to Bortezomib depends on the tumour cell type. Pancreatic, prostate and non-small lung tumour cells are sensitive while diffuse large B cell lymphoma, follicular lymphoma, Waldenstrom’s macroglobulinemia and some solid tumours such as sarcoma, renal cell carcinoma and glioma only provide slight and short responses (Frankland-Searby and Bhaumik 2012).
To overcome Bortezomib resistance, numerous combinations with chemotherapeutic agents, immunomodulators and/or other proteasome inhibitors are under investigation. In 2007, the FDA approved the use of a Bortezomib/Doxorubicin combination since it was better than the use of single compounds in phase III trials. Combinations with Doxorubicin/Adriamycin (ADR), Thalidomide, Melphalan, Dexamethasone, Cyclophosphamide and Myriad have also been successful. The most active combination to date includes Bortezomib, Lenalidomide and Dexamethasone, where overall responses of 100 % were obtained in phase II. Early phase I and II clinical trials using combinations of Bortezomib with new immunomodulators, radio-immunotherapy, stem cell transplantation, monoclonal antibodies or chemotherapeutic agents have been promising. In addition, targeted therapies such as histone deacetylase inhibitors, Bcl-2 inhibitors, rapamycin inhibitors, multiple kinase inhibitors (Akt, PKC, CDK inhibitors, etc.) and heat shock protein inhibitors simultaneously used with Bortezomib also show encouraging results (McBride and Ryan 2013).
MLN9708 shows synergistic effects in association with Bortezomib, vorinostat, lenalodomide and dexamethasone, in refractory MM (phase III) and first line MM patients (phase I/II) (McBride and Ryan 2013). Studies in phase I/II with Carfilzomib exhibit durable responses with acceptable toxicity in relapsed/refractory MM patients compared to Bortezomib and stem cell transplants, achieving a 24 % partial response in heavily pre-treated patients (McBride and Ryan 2013; Pautasso et al. 2013). Several phase II trials are currently on-going for haematological malignancies and solid tumours. Phase I/II trials with different compound combinations including Calfizomib/Thalidomide/Dexamethasone/Cyclosporamide are being also tested as a frontline treatment for MM, showing promising activity with high response rates in the first patients evaluated.
The last proteasome inhibitor that entered into phase I clinical trials was Marizomib, used alone or in combination with Dexamethasone for the treatment of different kinds of cancer (see Table 11.1). In preclinical trials, Marizomib is also combined with Bortezomib, since different mechanisms of action have been suggested (McBride and Ryan 2013).
11.3 Targeting NEDDylation
Together with ubiquitin and SUMO, NEDD8 is among the most studied ubiquitin-like proteins (Fig. 11.1). The best known physiological substrates of NEDDylation are the cullins, which are structurally related proteins that function as molecular scaffolds of the cullin-RING family of E3 ubiquitin-ligases (CRLs). CRLs consist of a core cullin protein bound to a RING finger protein (Rbx1/2) and an interchangeable substrate-binding adaptor protein (Bedford et al. 2011; Watson et al. 2011). Rbx1 and Rbx2 in conjunction with E2 enzymes UBC12 and UBE2F, respectively, promote NEDDylation of cullins (Huang et al. 2009). The archetypal CRLs, the so-called SCF ubiquitin ligases, contain CUL1, RBX1/2 and the adaptor protein Skp1. This core complex binds to one of the approximately 100 F-box proteins that are responsible for recruiting substrates (Bedford et al. 2011). Cullin NEDDylation has been shown to increase the ubiquitylation activity of CRLs by promoting conformational changes that increase the binding of Rbx1 to ubiquitin E2s, which results in a reduction of the distance between E2 and the substrate recognition component; it may also lead to displacement of the negative regulatory protein, CAND1, that binds to non-NEDDylated cullin (Watson et al. 2011). Specifically, the CRLs have been established to control the degradation of proteins with important biological roles, including cell cycle progression (p27, cyclin E, c-Myc), tumour suppression (p53), DNA damage (CDT1), stress responses (NRF-2, HIF-1α), and signal transduction (IκBα). In addition, other NEDD8-regulated substrates with key cancer-related functions are β-catenin, c-JUN, mTOR and MDM2. Interestingly, increasing evidence suggests that NEDD8-mediated protein turnover may be deregulated in malignant cells and could result in oncogenic transformation, disease progression, or impart a drug-resistant phenotype (Nawrocki et al. 2012). For example, disruptions in the NEDD8 pathway lead breast cancer cells to acquire anti-estrogen resistance and expression of ER alpha (Fan et al. 2003). It has also been shown that increased NEDD8 conjugation appeared to be essential for the enhancement of proliferation in several types of human carcinoma cells (Chairatvit and Ngamkitidechakul 2007).
11.3.1 NEDDylation Inhibitors
Because many CRL substrates have tumour suppressor activity, preventing the degradation of these proteins could be an effective anticancer strategy that may also help to reduce toxicities resulting from global proteasomal inhibition (Nawrocki et al. 2012). MLN4924, a potent and selective first-in-class small molecule inhibitor developed by Millennium Pharmaceuticals, was reported as a specific inhibitor of protein NEDDylation through the inactivation of the heterodimer APPBP1 (NAE1) and UBA3 (UBE1C), also known as the NEDD8 activating enzyme NAE (Soucy et al. 2009; Wang et al. 2011). MLN4924 is an adenosine sulphamate that forms a covalent adduct with NEDD8 when bound to the NAE active site and in this way inhibits the formation of UBC12-NEDD8 thioester. The MLN4924-NEDD8 adduct mimics the NEDD8-AMP in situ which is the first intermediate in the NAE reaction cycle, but cannot be enzymatically processed further (Brownell et al. 2010; Nawrocki et al. 2012). MLN4924 inhibits CRL activity and the stabilization of their substrates by blocking NAE, with subsequent implications in cancer cell growth and survival (Bedford et al. 2011). In 2009, MLN4924 was first reported as a potent growth suppressor agent against a variety of cancer cells lines derived from solid tumours and haematological malignancies in both in vitro and in vivo xenograft models (Soucy et al. 2009; Zhao et al. 2014). During preclinical trials, tumour regression occurs after the action of different mechanisms depending on context and cell type. Induction of apoptosis is one of the reported effects mediated by MLN4924 in three different mechanisms mainly caused by the accumulation and stabilization of CRL substrates like: (1) CDT1, which trigger DNA re-replication and S phase arrest; (2) IκBα, that blocks NF-κB activation, and; (3) pro-apoptotic NOXA. In addition to apoptosis, MLN4924 also induces autophagy in a concentration- and time-dependent manner in multiple human cancer cell lines derived from carcinomas of breast, colon, liver, brain and cervix. Studies revealed that autophagy is mainly caused by inactivation of mTORC1, most likely mediated by accumulation of DEPTOR and HIF1α, followed by the activation of the HIF1-REDD1-TSC1 axis. Some studies have also shown that MLN4924 can induce irreversible senescence in multiple cancer cell lines in a p21-dependent manner. In SK-BR3 cells, p16 and p27 accumulation may also contribute to the MLN4924-induced senescence. Furthermore, MLN4924 has a potential sensitizer role in chemotherapy and radiation, involving the mechanistic accumulation of c-Jun, NOXA, CDT1, WEE1 or p21; it also affects the promotion of c-FLIP degradation, increasing the expression of Bcl-2-interacting killer (BIK), inactivating CRL3, as well as the suppression of FANCD2 monoubiquitylation and CHK1 phosphorylation, all of which results in a general increase in cellular sensitivity by enhancing DNA damage, oxidative stress, cell cycle arrest and finally apoptosis (Zhao et al. 2014). Despite this promising optimal strategy to inhibit NAE pathway, recently it has been identified that cancer cells can develop resistance to MLN4924 treatment. Resistance has been linked to heterozygous mutations in two areas of NAEβ (UBA3), the ATP binding pocket at Alanine 171 and at various residues within or close to the NEDD8-binding cleft. A point mutation in residue A171T reduced affinity for both MLN4924 and ATP. Interestingly, such resistance has been effectively bypassed by using a compound with tighter binding properties for NAE. These findings provide critical clinical aspects with respect to patient selection and consolidate efforts for the development of next-generation NAE inhibitors in order to overcome emergent mutations (Milhollen et al. 2012; Toth et al. 2012). In fact, by using a virtual screening approach, a natural product, 6,6″-biapigenin, has been identified and proposed as a second inhibitor of NAE (Leung et al. 2011). In 2012, a cyclometallated rhodium (III) complex was reported as the first metal complex to suppress the NEDDylation pathway via inhibition of NAE, which occupied the same binding pocket as MLN4924 (Zhong et al. 2012).
11.3.2 Experience with Clinical Trials
In parallel with the promising results in preclinical models, MLN4924 has been included in clinical trials for cancer therapy since 2008 (see Table 11.2). Up to now, there are a total of seven Phase I/II clinical trials for MLN4924 in patients with leukaemia, lymphoma, melanoma and several solid tumours. These trials were mainly designed to assess the safety, discomforts and risks of the inhibitor; to establish the maximum tolerated dose (MTD) that can be given to patients; to describe the pharmacokinetics (PK) parameters and pharmacodynamics (PD) effects; to evaluate disease response, and to study MLN4924 in combination with other standard treatments. In general, patients received escalating doses of MLN4924 that was administrated via intravenous infusion on different daily schedules. The pharmacokinetics of MLN4924 were measured in serial blood samples, bone marrow aspirates (BMAs), skin punch biopsies, or fine-needle tumour biopsies collected following drug dosing. The samples were analysed to measure MLN4924-NEDD8 covalent adduct and the expression of CRL substrates, such as CDT1, NRF2, and phospho-IκBα, as the biomarkers to indicate NAE inhibition in peripheral and tumour tissue (Zhao et al. 2014). The PK profiles were similar following dosing on different days, suggesting no apparent accumulation of MLN4924 in plasma with an estimated half-life of 5–15 h. In general, most clinical trials carried out thus far have concluded that MLN4924 is well tolerated in the majority of the dosing schedules studied, with evidence of target inhibition and antitumoural activity which supports continued investigation of MLN4924, both alone and in combination strategies as potential treatment for a variety of human cancers (Nawrocki et al. 2012; Zhao et al. 2014).
Table 11.2
Clinical trials for MLN4924
Disease type | Clinical status | With combination | MTD | Responses | Ref |
|---|---|---|---|---|---|
Advanced Solid tumors | Phase I | Alone With dexamethasone (Dex) | 50 mg/m2 in Schedule A (days 1-5); | Stable disease | Clinicaltrials.gov Identifier: NCT00677170 Kauh et al. 2011. J Clin Oncol (ASCO Annual Meeting Abstracts) 29 (suppl; abstr 3013) |
67 mg/m2 in Schedule B (days 1, 3 and 5 + Dex); | |||||
50 mg/m2 in Schedule C (days 1, 3 and 5) | |||||
Relapsed and/or refractory Hodgkin’s Lymphoma, Non-Hodgkin’s Lymphoma and Multiple Myeloma | Phase I | Alone | 110 mg/m2 in Schedule A (days 1, 2, 8 and 9) | 3 partial response and stable disease | Clinicaltrials.gov Identifier: NCT00722488 (Nawrocki et al. 2012) Shah et al. 2010. Blood (ASH Annual Meeting Abstracts) 116: abstract 2801 Harvey et al. 2012 (17th Congress of European Hematology Association) abstract 1060 TrialTroveID-170665 |
Not reported for Schedule B (days 1,4, 8 and 11) and Schedule C (days 1 and 8) | |||||
196 mg/m2 (twice-weekly schedule of days 1, 4, 8 and 11) | |||||
Acute Myeloid Leukemia, Acute Lymphoblastic Leukemia, Myelodysplastic Syndromes | Phase I | Alone With azacitidine | 59 mg/m2 (days 1, 3 and 5) | 4 complete responses | Clinicaltrials.gov Identifier: NCT00911066 Swords et al. Blood (ASH Annual Meeting Abstracts) 116: abstract 658 Nawrocki et al. (2012) Zhao et al. (2014) |
Metastatic Melanoma | Phase I | Alone | Has not been defined | 1 partial response and 9 stable disease | Clinicaltrials.gov Identifier: NCT01011530 Bhatia et al. 2011. J Clin Oncol (ASCO Annual Meeting Abstracts) 29 (suppl; abstr 8529) |
Large B-cell Lymphoma | Phase I/II | Alone With standard EPOCH-R chemotherapy | Not reported | Not reported (study was withdrawn before participants were enrolled) | Clinicaltrials.gov Identifier: NCT01415765 Zhao et al. (2014) |
Acute Myeloid Leukemia | Phase Ib | Alone With azacitidine | To be determined | Not reported | Clinicaltrials.gov Identifier: NCT01814826 Zhao et al. (2014) |
Solid tumors | Phase Ib | With docetaxel With gemcitabine With carboplatin + paclitaxel | To be determined | Not reported | Clinicaltrials.gov Identifier: NCT01862328 Zhao et al. (2014) |
11.4 Targeting DUBs
The human genome encodes nearly 100 DUBs in five major classes: 60 Ubiquitin-specific proteases (USPs), 16 Otubain-domain containing proteins (OTUs), 4 Machado-Joseph Domain (Josephin domain)-containing proteins (MJD), 4 ubiquitin C-terminal hydrolases (UCHs) and 8 Jab1/MPN domain-associated metalloisopeptidases (JAMM). USPs, OTUS, MJD and UCHs use an active site cysteine as a nucleophile to attack lysine-glycine isopeptide bonds within ubiquitylated proteins (Nijman et al. 2005), whereas the fifth class of DUBs contains a JAMM zinc metalloproteinase domain (Cope and Deshaies 2003). DUBs are generally expressed as active enzymes, rather than inactive precursors. However, certain DUBs require ubiquitin binding to obtain their active conformation and prevent their uncontrolled proteolytic activity. Structural data revealed that ubiquitin-binding by DUBs is accompanied by active site rearrangements, which are necessary to induce their hydrolytic activity (Edelmann et al. 2009; Komander et al. 2009). DUB activity is also regulated through the binding of scaffold and adaptor proteins (Ventii and Wilkinson 2008), proteolytic cleavages, as well as post-translational modifications (Kessler and Edelmann 2011).
11.4.1 DUBs as Therapeutic Targets
Due to the implication of DUBs in the regulation of crucial signalling pathways such as p53 and NF-κB (Fig. 11.2), it is not surprising that their deregulation is involved in a growing number of diseases, including neurological disorders, viral infections and cancer. Indeed, some members of the DUBs family are known to contribute to neoplastic transformation, such as USP1 (in Fanconi anaemia), USP2 (in prostate cancer), DUB3 (in breast cancer), USP4 (in adenocarcinoma), USP7 (in prostate cancer and non-small-cell lung adenocarcinoma), USP9X (in both leukaemia and myelomas) and BRCC36 (in breast cancer) (Edelmann et al. 2011; Hussain et al. 2009). The genetic alteration of DUBs such as CYLD and USP6, has been associated with skin and bone marrow tumour progression, respectively. Finally, an alteration of expression of A20 (B-cell and T-cell lymphomas), USP10 (carcinomas) and BAP1 (brain, lung and testicular cancers) is observed in some cancers (Hussain et al. 2009; Nicholson et al. 2007; Sippl et al. 2011).
The high degree of substrate specificity and the well defined catalytic pocket make DUBs druggable and amenable to screening with libraries of small molecules (Eletr and Wilkinson 2014). Work carried out over the last few years has led to the identification of inhibitors with selective action against various USP targets, demonstrating the feasibility of selective targeting of DUBs. In general, the initial hits have been obtained by high throughput screening (HTS) of compound libraries, followed by further optimization using structure-activity relationships (Kramer et al. 2012). Companies like Novartis, Progenra and Hybrigenics have patented compounds inhibiting distinct DUBs (see Table 11.3). Some of them will be presented in the following section. However, to the best of our knowledge, only a few of them have, as yet, entered into clinical trials.
Table 11.3
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells




Inhibitors of Deubiquitylating enzymes in clinical trials
Related posts:
Introduction to Cell Stress Responses in Cancer: The Big Picture
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Inflammatory Dysregulation and Cancer: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
The Hsp70 Family of Heat Shock Proteins in Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
