The Thyroid Gland: Introduction
The thyroid gland is the body’s largest single organ specialized for endocrine hormone production. Its function is to secrete an appropriate amount of the thyroid hormones, primarily 3,5,3′,5′-l-tetraiodothyronine (thyroxine, T4), and a lesser quantity of 3,5,3′-l-triiodothyronine (T3), which arises mainly from the subsequent extrathyroidal deiodination of T4. In target tissues, T3 interacts with nuclear T3 receptors that are, in turn, bound to special nucleotide sequences in the promoter regions of genes that are positively or negatively regulated by thyroid hormone. Among their life-sustaining actions, the thyroid hormones promote normal fetal and childhood growth and central nervous system development; regulate heart rate and myocardial contraction and relaxation; affect gastrointestinal motility and renal water clearance; and modulate the body’s energy expenditure, heat generation, weight, and lipid metabolism. In addition, the thyroid contains parafollicular or C cells that produce calcitonin, a 32-amino-acid polypeptide that inhibits bone resorption, but has no apparent physiologic role in humans. However, calcitonin is clinically important as a tumor marker produced by medullary thyroid cancers that arise from these cells (Chapter 8).
Embryology, Anatomy, and Histology
The thyroid gland originates in the embryo as a mesodermal invagination in the pharyngeal floor at the foramen cecum, from which it descends anterior to the trachea and bifurcates, forming two lateral lobes, each measuring approximately 4 cm in length, 2 cm in width, and 1 cm in thickness in adulthood. Ectopic thyroid tissue can be present anywhere along or beyond this thyroglossal duct, from the tongue base (lingual thyroid) to the mediastinum. The thyroglossal duct may also give rise to midline cysts lined with squamous epithelium, which can remain asymptomatic, or become infected or give rise to thyroid tumors. The caudal end of the thyroglossal duct forms the pyramidal lobe of the thyroid, which can become palpable in conditions causing diffuse thyroid inflammation or stimulation (Figure 7–1).
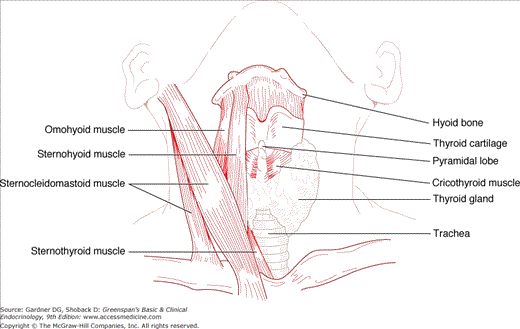
Upward growth of the thyroid gland is limited by the attachment of the sternothyroid muscle to the thyroid cartilage. However, posterior and downward growth is unimpeded, so that thyroid enlargement, or goiter, frequently extends posteriorly and inferiorly, even into the superior mediastinum (substernal goiter).
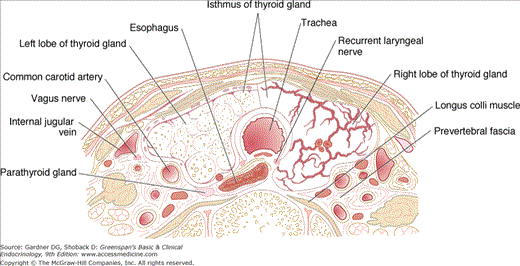
The thyroid gland has clinically important anatomical relationships to the recurrent laryngeal nerves, which course behind the gland, and two pairs of parathyroid glands that usually lie behind the upper and middle portions of the thyroid lobes. The thyroid is also draped around the trachea and the posterior margins of its lobes abut the esophagus. All of these structures can be compressed by gland enlargement, invaded by thyroid malignancies, or injured in the course of thyroid surgery (Figure 7–2). Because the posterior thyroid capsule is bound to the pretracheal fascia, the gland normally rises and falls with deglutition, facilitating its inspection and palpation.
The thyroid gland has a rich blood supply (Figure 7–3), which can be increased in hyperthyroidism, giving rise to an audible whooshing sound (bruit) or even a palpable vibration (thrill). Microscopically, thyrocytes form hollow spheres (follicles) that surround a central lumen containing an aggregation of iodinated thyroglobulin (colloid) that represents the gland’s hormone stores (Figure 7–4).
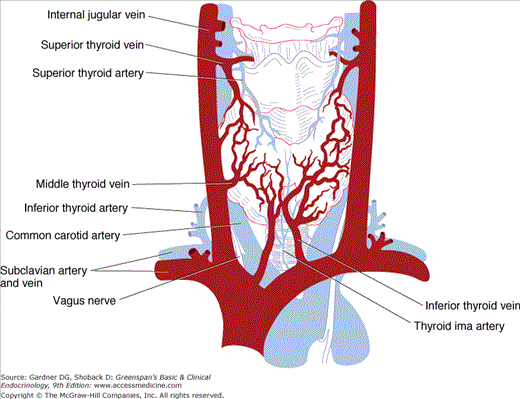
Figure 7–4
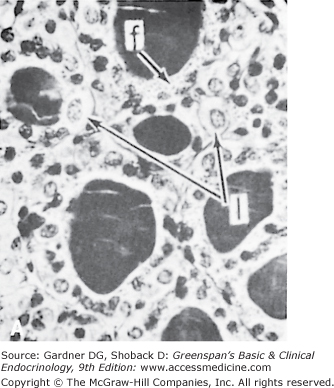
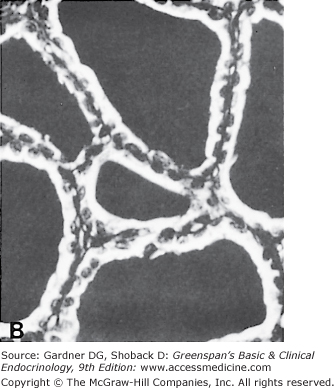
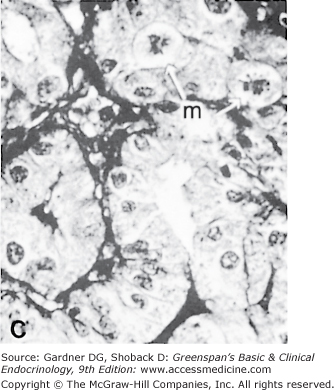
A. Normal rat thyroid. A single layer of cuboidal epithelial cells surrounds PAS-positive material in the follicular space (colloid). The larger, lighter-staining cells indicated by the arrows (I) are C cells that produce calcitonin. The follicular cells form an epithelial layer that surrounds the colloid (F). B. Inactive rat thyroid several weeks after hypophysectomy. The follicular lumens are larger and the follicular cells flatter. C. Rat thyroid under intensive TSH stimulation. The animal was fed an iodine-deficient diet and injected with propylthiouracil for several weeks. Little colloid is visible. The follicular cells are tall and columnar. Several mitoses (m) are visible.
(Reproduced, with permission, from Halmi NS. In: Greep RO, Weiss L, eds. Histology. McGraw-Hill; 1973.)
Physiology
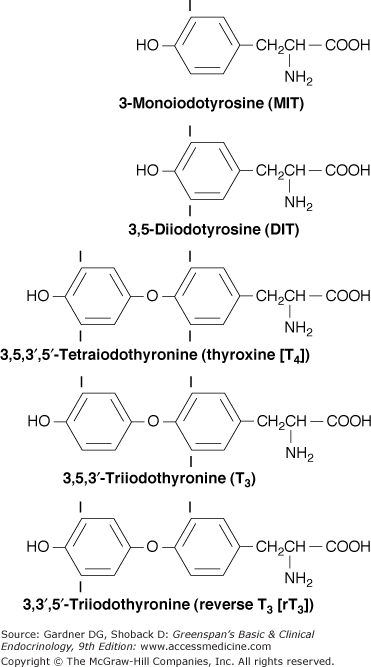
The thyroid hormones are iodinated thyronines, which comprise of two tyrosine moieties joined by an ether linkage (Figure 7–5). The follicular cells of the thyroid gland are specialized in their ability to synthesize the large hormonal precursor protein thyroglobulin, concentrate iodide intracellularly from the circulation, and express a receptor that binds thyroid-stimulating hormone (thyrotropin, TSH), which promotes thyrocytes’ growth and biosynthetic functions.
Iodine Metabolism
Iodine1 is a key structural component of thyroid hormones. Consequently, it is an essential micronutrient consumed in food or water as iodide or iodate, which is converted to iodide in the stomach. The World Health Organization (WHO) recommends a daily dietary iodine intake of 150 μg for adults, 200 μg for pregnant and lactating women, and 50 to 250 μg for children. Because most iodide is excreted by the kidneys, urinary iodide excretion is an excellent index of dietary intake.
Over millennia, iodine has been leached from the soil in many mountainous and inland regions of the world. Consequently, according to WHO, dietary iodine deficiency, defined as a daily iodine intake less than 100 μg/d, affects an estimated two billion people, which is about one-third of the world’s population. When iodide intake is less than 50 μg/d, a normal-sized thyroid cannot sustain adequate hormone production, with resulting gland enlargement (goiter) and, ultimately, hypothyroidism. The consequences of endemic dietary iodine deficiency are especially devastating for the developing fetus and children, who require thyroid hormone for normal neurologic development and growth. In the North American diet, iodine is principally derived from iodized salt, iodate preservatives in baked goods, dairy products containing traces of iodophore antibacterial agents used in milk collection, food coloring, and seafood.
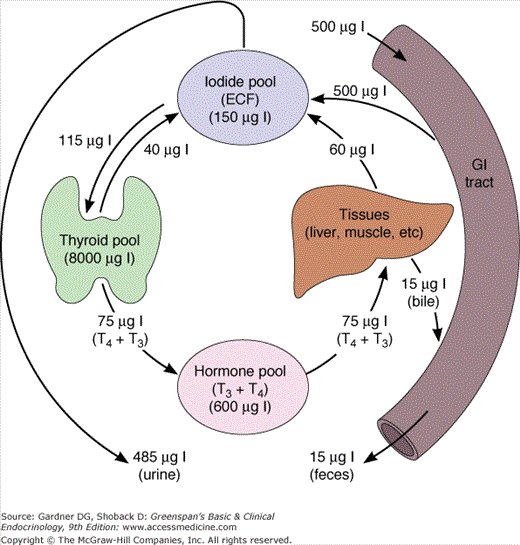
Thyrocytes abundantly express the sodium-iodide symporter (Na-I− symporter; NIS), which spans the cells’ basal membranes and actively transports iodide from the blood. The thyroid gland concentrates and uses only a fraction of the iodide supplied to it for hormone synthesis, and the remainder returns to the extracellular fluid pool. Consequently, the normal fractional uptake of iodide, which can be quantified with a radioactive iodine tracer, is approximately 10% to 30% after 24 hours. Because of this active concentrating mechanism and the subsequent organification of intracellular iodide, the intrathyroidal pool of iodine is very large, 8 to 10 mg in the form of stored thyroid hormones and iodinated tyrosines. This iodine provides a buffer in the event of temporary dietary iodine deficiency.
Synthesis of T4 and T3 by the thyroid gland involves six major steps: (1) active transport of iodide across the basement membrane into the thyroid cell (trapping); (2) oxidation of iodide and iodination of tyrosyl residues in thyroglobulin (organification); (3) linking pairs of iodotyrosine molecules within thyroglobulin to form the iodothyronines T3 and T4 (coupling); (4) pinocytosis and then proteolysis of thyroglobulin with release of free iodothyronines and iodotyrosines into the circulation; (5) deiodination of iodotyrosines within the thyroid cell, with conservation and reuse of the liberated iodide; and (6) intrathyroidal 5′-deiodination of T4 to T3.
Thyroid hormone synthesis requires that NIS, thyroglobulin, and the enzyme thyroid peroxidase (TPO) all be present, functional, and uninhibited. This process is summarized in Figures 7–6 and 7–7.
Figure 7–6
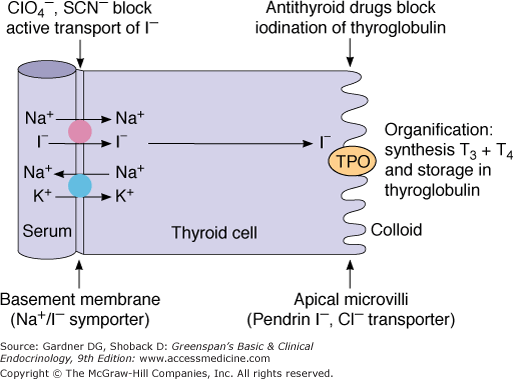
The iodide transporter in the thyroid cell. The large solid circle represents the Na+/I− symporter actively transporting I− into the cell; the large blue circle represents Na+-K+ ATPase supplying the ion gradient which drives the reaction. I− is transported across the apical membrane by pendrin. Hormone synthesis takes place in the colloid at the colloid-apical membrane, catalyzed by thyroperoxidase (TPO).
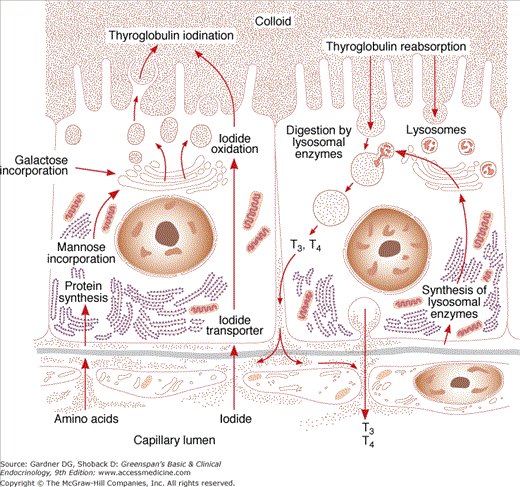
Thyroglobulin is a large glycoprotein molecule (MW 660,000 kD), composed of two subunits, each containing 5496 amino acids. Thyroglobulin includes approximately 140 tyrosyl residues, but only four tyrosyl sites are sterically oriented for effective hormonogenesis in each molecule. The iodine content of thyroglobulin can vary from 0.1% to 1% by weight. In thyroglobulin containing 0.5% iodine, for example, there are approximately three molecules of T4 and one molecule of T3.
The thyroglobulin gene, which resides on the long arm of chromosome 8, contains approximately 8500 nucleotides, which encode the prethyroglobulin protein monomer, including a 19-amino acid signal peptide. TSH regulates expression of the thyroglobulin gene. After thyroglobulin mRNA is translated in the rough endoplasmic reticulum (RER), the protein is glycosylated during transport through the Golgi apparatus (see Figure 7–7), where thyroglobulin dimers are incorporated into exocytic vesicles. These vesicles then fuse with the cell’s apical basement membrane, from which they are released into the follicular lumen. There, at the apical-colloid border, tyrosine residues in thyroglobulin are iodinated.
Iodide (I−) is transported across thyrocytes’ basal membrane by the NIS. Membrane-bound NIS, which derives its energy from a Na+-K+ ATPase, allows the human thyroid gland to maintain a concentration of free iodide 30 to 40 times higher than that in plasma. NIS action is stimulated physiologically by TSH and pathophysiologically by the TSH receptor–stimulating antibody of Graves disease. Although salivary, gastric, and breast tissues express NIS and concentrate iodide to a lesser extent than the thyroid, these other tissues do not organify or store iodide, and their NIS activities are not stimulated by TSH. Large amounts of iodide suppress both NIS activity and NIS gene expression, representing mechanisms of iodine autoregulation (see later). The perchlorate ion (ClO4−) competes with iodide for NIS; perchlorate has been used to treat hyperthyroidism and has the potential to be an environmental inhibitor of thyroid function. NIS can also concentrate pertechnetate (TcO4−) into thyroid cells, facilitating use of the radionuclide sodium pertechnetate (Tc99mO4−) for visualization of the thyroid gland and quantification of its trapping activity.
At the thyrocyte’s apical border, a second iodide transport protein, pendrin, transports iodide to the membrane–colloid interface, where it becomes a substrate for thyroid hormonogenesis (see Figures 7–6 and 7–7). Mutations in the pendrin (PDS or SLC26A4) gene impairing its function cause a syndrome of goiter and hearing loss acquired in infancy or early childhood (Pendred syndrome).
Thyroid peroxidase (TPO), a membrane-bound glycoprotein (MW 102 kD) containing a heme moiety, catalyzes both iodide oxidation and covalent linkage of iodine to the tyrosine residues of thyroglobulin. TPO gene expression is stimulated by TSH. After TPO is synthesized in the RER, it is inserted into the RER’s cisternal membranes, from which it is transferred to the apical cell surface through Golgi elements and exocytic vesicles. Here, at the cell–colloid interface, TPO facilitates both iodination and coupling of the tyrosine residues within thyroglobulin.
Within the thyroid cell, at the apical–colloid interface, iodide is rapidly oxidized by locally produced hydrogen peroxide in a reaction catalyzed by TPO; the resulting active iodide intermediate is bound to tyrosyl residues in thyroglobulin. The required hydrogen peroxide is probably generated by an NADPH oxidase in the presence of calcium cations, a process that is also stimulated by TSH. TPO can also catalyze iodination of tyrosyl molecules in proteins other than thyroglobulin, such as albumin or thyroglobulin fragments; but no active hormones arise from these proteins.
The coupling of iodotyrosyl residues in thyroglobulin is also catalyzed by TPO. This is believed to be an intramolecular process involving oxidation of two iodotyrosyl residues brought into proximity by the tertiary and quaternary structures of thyroglobulin, their linkage as a quinol ether intermediate, and splitting of the quinol ether to form an iodothyronine (Figure 7–8). Within the thyroglobulin molecule, two molecules of diiodotyrosine (DIT) couple to form T4, and monoiodotyrosine (MIT) and DIT molecules couple to form T3.
Figure 7–8
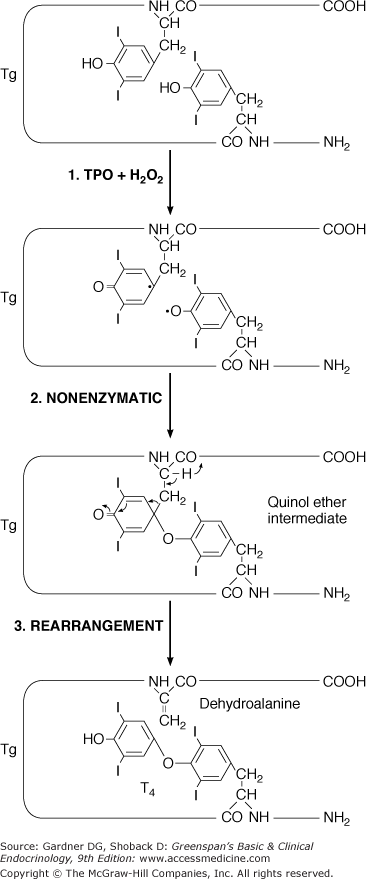
Hypothetical coupling scheme for intramolecular formation of T4 within the thyroglobulin molecule. The major hormonogenic site at tyrosyl residue 5 is indicated.
(Reproduced, with permission, from Taurog A. Thyroid hormone synthesis. Thyroid iodine metabolism. In Braverman LE, Utiger RD, eds. Werner and Ingbar’s The Thyroid. 7th ed. Lippincott; 1996.)
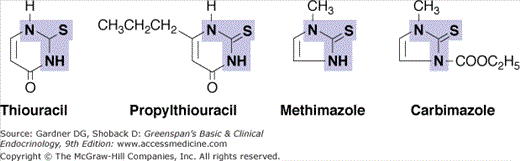
The thiocarbamide drugs, including methimazole, carbimazole, and propylthiouracil (PTU), are competitive inhibitors of TPO. Their resulting ability to block thyroid hormone synthesis (Figure 7–9) makes them useful in treatment of hyperthyroidism.
The processes of thyroglobulin proteolysis and secretion in thyroid hormonogenesis are illustrated in Figure 7–7. At the thyrocyte’s apical membrane, colloid is engulfed in vesicles by pinocytosis and absorbed into the cell. Lysosomes containing proteolytic enzymes then fuse with the colloid vesicle. This releases T4 and T3, as well as inactive iodotyrosines, peptides, and individual amino acids. The biologically active thyroid hormones T4 and T3 enter the circulation; DIT and MIT are deiodinated and their iodide conserved. Thyroid hormone secretion is stimulated by TSH and inhibited by excess iodide (see later) and lithium. A small amount of intact thyroglobulin is normally released from thyroid cell and circulates in blood. The serum thyroglobulin concentration is markedly increased in a number of thyroid conditions, including thyroiditis, nodular goiter, and Graves disease. Because thyroglobulin is also synthesized by most malignancies arising from thyroid epithelium, such as papillary and follicular thyroid cancers, it is a useful circulating tumor marker.
MIT and DIT formed during the synthesis of thyroid hormone are deiodinated by intrathyroidal deiodinase, an NADPH-dependent flavoprotein found in mitochondria and microsomes that acts on the iodotyrosines MIT and DIT, but not on T3 and T4. Most of the iodide released is reused for hormone synthesis, and only a small amount normally leaks out of the thyroid gland (Figure 7–10).
The 5′-deiodinase that converts T4 to T3 in peripheral tissues is also found in the thyroid gland. When there is iodide deficiency and in various hyperthyroid states, the activity of this enzyme increases the amount of T3 secreted by the gland, increasing the metabolic efficiency of hormone synthesis.
A very low iodine diet and inherited defects in genes encoding the proteins required for thyroid hormone biosynthesis (dyshormonogenesis) can both result in insufficient hormone production. In response to limited intrathyroidal iodine content or hormone production, the gland increases the ratios of MIT-to-DIT within thyroglobulin as well as the proportion of secreted T3 relative to T4. The hypothalamic-pituitary-thyroid axis also responds to thyroid hormone deficiency by increasing TSH secretion. Consequently, affected individuals typically present with thyroid gland enlargement (goiter), which may be sufficient to compensate for inefficient thyroid hormone production; but if not, they develop hypothyroidism. Severely affected neonates and infants can suffer the irreversible effects of thyroid hormone deficiency on development that result in cretinism. Specific inherited disorders are described in more detail in the section on nontoxic goiter, below.
Although iodide is an essential substrate for thyroid hormone production, excess iodide actually inhibits three steps in thyroid hormone production: iodide trapping, thyroglobulin iodination (the Wolff-Chaikoff effect), and thyroid hormone release from the gland. These inhibitory actions are transient, and the normal thyroid gland escapes after 10 to 14 days from these effects of excess iodide. These autoregulatory effects of iodide insulate physiologic thyroid function from short-term fluctuations in iodine intake.
These actions of excess iodide also have important clinical implications, sometimes causing iodine-induced thyroid dysfunction and permitting iodide treatment of certain thyroid conditions. If the thyroid is affected by autoimmune thyroiditis or certain inherited forms of dyshormonogenesis, it may be incapable of escaping from iodide-induced inhibition of gland function, and hypothyroidism can develop. Conversely, an iodide load can induce hyperthyroidism (Jod-Basedow effect2) in some patients with multinodular goiter, latent Graves disease, and rarely in individuals with thyroid glands that appear otherwise normal.
Both thyroid hormones circulate in blood bound to plasma proteins; only 0.04% of T4 and 0.4% of T3 are unbound or free, and consequently, available for entry and action in target tissues (Figure 7–11). There are three major thyroid hormone transport proteins: thyroxine-binding globulin (TBG); transthyretin, formerly called thyroxine-binding prealbumin (TBPA), and albumin (Figure 7–12). The plasma protein binding permits blood delivery of the iodothyronines, which are otherwise poorly soluble in water. It also creates a large circulating thyroid hormone pool with a stable 7-day plasma half-life and ensures the homogeneous distribution of thyroid hormones in target tissues.
Figure 7–11
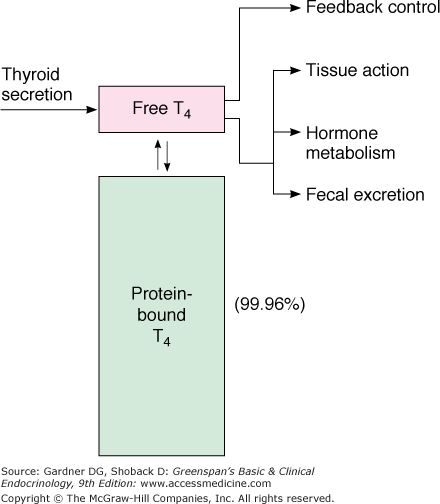
Representation of free T4 (and free T3) as the biologically active hormones at the level of the pituitary and the peripheral tissues. Most of the thyroid hormones circulating in plasma are protein-bound and have no biologic activity. This pool of bound hormone is in equilibrium with the free hormone pool.
(Reproduced, with permission, from DeGroot LJ, Stanbury JB. The Thyroid and Its Diseases. 4th ed. Wiley; 1975.)
Figure 7–12
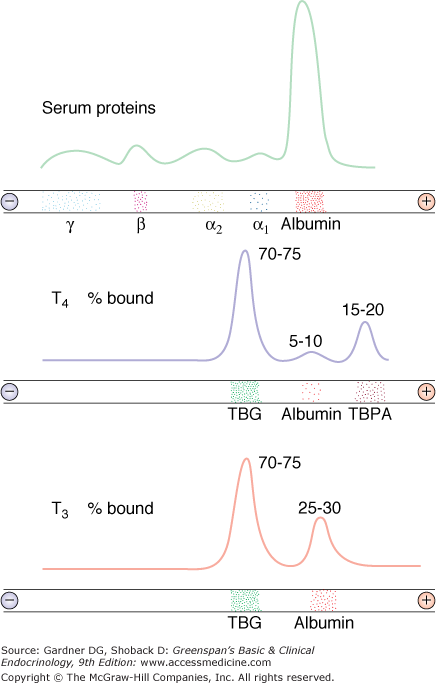
Diagrammatic representation of the distribution of radioactive T4 and T3 among serum thyroid hormone–binding proteins. Top: Paper electrophoretic pattern of serum proteins. Middle: Radioactive T4 was added to serum and was then subjected to paper electrophoresis. The peaks represent the mobility of radioactive T4 bound to different serum proteins. (TBG, thyroid hormone–binding globulin; TBPA, thyroxine-binding prealbumin.) Bottom: Radioactive T3 was added to serum and subjected to paper electrophoresis. The peaks indicate the relative distribution of protein-bound radioactive T3. The figures above each peak indicate the relative hormone distribution among the binding proteins in a normal adult.
(Reproduced, with permission, from Rosenfield RL, et al. Pediatric Nuclear Medicine. James AE Jr, Wagner HN Jr, Cooke RE, ed. Saunders; 1974.)
Thyroxine-binding globulin (TBG) is a liver-derived glycoprotein member of the SERPIN family of serine antiproteases composed of a single 54-kDa polypeptide chain, to which are attached 4 carbohydrate chains normally containing approximately 10 sialic acid residues. Each TBG molecule has a single binding site for T4 or T3. The serum concentration of TBG is 15 to 30 μg/mL (280-560 nmol/L), and its high binding affinity for T4 and T3 allows it to carry about 70% of circulating thyroid hormones.
TBG and its binding of the thyroid hormones can be altered by congenital TBG derangements, certain physiologic and pathophysiologic circumstances, and several drugs. TBG deficiency occurs with a frequency of 1:5000 live births, with a number of variants described in several ethnic and racial groups. It is an X-linked recessive trait that, consequently, is much more commonly expressed in males. Despite low circulating total T4 and T3 levels in affected individuals, free hormone levels are normal, and these patients remain euthyroid. Congenital TBG deficiency is often associated with congenital corticosteroid-binding globulin deficiency (Chapter 9). Conversely, congenital TBG excess, which is rare, is characterized by elevated total T4 and T3 concentrations in blood, but normal free hormone levels and a euthyroid clinical state. Pregnancy, estrogen-secreting tumors, and estrogen therapy all increase the sialic acid content of the TBG molecule, resulting in decreased metabolic clearance and elevated serum TBG levels. TBG levels can decrease in major systemic illness, and due to cleavage by leukocyte proteases, its binding affinity for the thyroid hormones can also be reduced. Both effects lower the serum total thyroid hormone concentrations in sick patients (Table 7–1). Certain drugs can also decrease (androgenic steroids, glucocorticoids, danazol, L-asparaginase) or increase (estrogens, 5-fluourouracil) the plasma TBG concentration. Other drugs (eg, salicylates, high-dose phenytoin, and intravenous furosemide) can bind to TBG, displacing T4 and T3. In this circumstance, the hypothalamic-pituitary-thyroid axis (discussed later) preserves normal, free hormone concentrations by lowering serum total thyroid hormone levels. Similarly, heparin stimulation of lipoprotein lipase releases free fatty acids that displace thyroid hormones from TBG. In vivo, this can result in lower total thyroid hormone concentrations, whereas in vitro (eg, in blood drawn through a heparin lock), heparin increases measured levels of free T4 and T3.
Euthyroid Hyperthyroxinemia
↓T4-to-T3 conversiona
Thyroxine therapy in hypothyroidisma Generalized resistance to thyroid hormonea
|
Euthyroid Hypothyroxinemia ↓Thyroxine-binding globulin
Systemic illnessb Medications Exogenous thyromimetic compounds (T3 [Cytomel])b
|
Transthyretin, a 55-kDa globular polypeptide composed of four identical 127-amino acid subunits, binds 10% of circulating T4. Its affinity for T4 is 10-fold greater than for T3. The dissociation of T4 and T3 from transthyretin is rapid, so that transthyretin is a source of readily available T4. Increased affinity of transthyretin binding for T4 can occur as a heritable condition. Affected individuals have an elevated total T4 but a normal free T4. Ectopic production of transthyretin, which has been reported to occur in patients with pancreatic and hepatic tumors, also causes euthyroid hyperthyroxinemia.
Albumin binds to T4 and T3 with lesser affinity than TBG or transthyretin, but its high plasma concentration results in its transport of 15% of circulating T4 and T3. Rapid thyroid hormone dissociation rates from albumin make it a major source of free hormone to tissues. Hypoalbuminemia, as occurs in nephrosis or cirrhosis, is associated with a low total T4 and T3, but the free hormone levels are normal.
Familial dysalbuminemic hyperthyroxinemia is an autosomal dominant inherited disorder in which 25% of the albumin exhibits a higher than normal T4-binding affinity. This results in an elevated total T4 level but a normal free T4 concentration and euthyroidism. In most affected kindreds, the T3-binding affinity is normal. Because these albumin variants do not bind the thyroxine analogs used in many free T4 immunoassays, they may falsely report elevation of the free T4 in affected individuals.
The normal thyroid gland secretes about 100 nmol of T4 and only 5 nmol of T3 daily; less than 5 nmol of metabolically inactive reverse T3 (rT3) is produced (Figure 7–13). Most of the plasma pool of T3 (80%) is derived from peripheral outer ring- or 5′-monodeiodination of T4 in tissues outside of the thyroid gland, particularly the liver, kidney, and skeletal muscle (Table 7–2). Because T3 has a higher binding affinity for the nuclear T3 receptors that affect thyroid hormone action, this 5′-monodeiodination generates a more biologically active iodothyronine. On the other hand, deiodination of the inner ring of T4 (5-deiodination) produces 3,3′,5′-triiodothyronine or reverse T3 (rT3), which is metabolically inert.
Figure 7–13
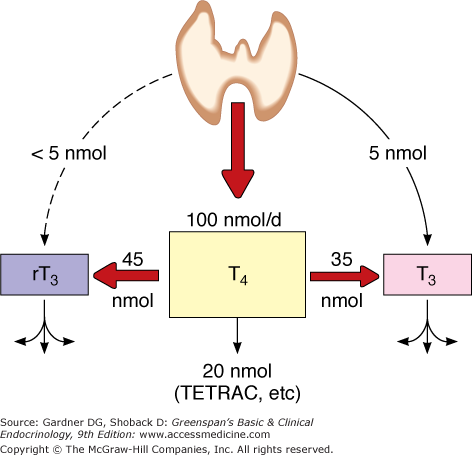
Major pathways of thyroxine metabolism in normal adult humans. Rates are expressed in nmol/24 h and are approximations based upon available data. 100 nmol of T4 is equivalent to approximately 75 μg. (rT3, reverse T3; TETRAC, tetraiodothyroacetic acid.)
(Reproduced, with permission, from Cavalieri RR, Rapoport B. Impaired peripheral conversion of thyroxine to triiodothyronine. Ann Rev Med. 1977;28:5765.)
| Deiodinase Type | D1 | D2 | D3 |
|---|---|---|---|
| Substrates | rT3 > T4 > T3 | T4 > rT3 | T3 > T4 |
| Tissue distribution | Liver, kidney, skeletal muscle, thyroid | Brain, pituitary | Brain, placenta, fetal tissues |
| Function | Plasma T3 production | Local T3 production | T3 degradation |
| PTU inhibition (IC50, μM) | 5 | > 1000 | > 1000 |
| Hypothyroidism | Decrease | Increase | Decrease |
| Hyperthyroidism | Increase | Decrease | Increase |
The three deiodinase enzymes that catalyze these reactions differ in tissue localization, substrate specificity, and physiologic and pathophysiologic modulation, as summarized in Table 7–2. Type 1 5′-deiodinase, the most abundant form, is found predominantly in liver and kidney, and in lesser quantities in the thyroid gland, skeletal and heart muscle, and other tissues. The type 1 5′-deiodinase contains a selenocysteine moiety that is probably at the active deiodinating site. The major function of type 1 5′-deiodinase is to provide T3 to the circulation. Its activity is increased in hyperthyroidism and decreased in hypothyroidism. This partially accounts for the relatively higher circulating T3 levels compared to T4 levels seen in hyperthyroid patients. The enzyme is inhibited by the thionamide antithyroid drug PTU, but not methimazole, and by the antiarrhythmic drug amiodarone and iodinated radiocontrast dyes, such as sodium ipodate. Dietary selenium deficiency can also impair T4 to T3 conversion.
Type 2 5′-deiodinase is predominantly expressed in the brain and pituitary gland, where it maintains a constant level of intracellular T3 in the central nervous system. This deiodinase is very sensitive to circulating T4, so a lower circulating T4 rapidly increases enzyme concentration in brain and pituitary, by altering the rate of T4 degradation and inactivation. In doing so, it maintains the level of intracellular T3 and its neuronal cellular functions. Conversely, elevated plasma T4 reduces the type 2 5′-deiodinase level, protecting brain cells from excessive T3. Consequently, this deiodinase represents a mechanism by which the hypothalamus and pituitary can respond to the level of circulating T4. rT3 can also modify the activity of type 2 5′-deiodinase in the brain and the pituitary gland, and α-adrenergic compounds stimulate type 2 5′-deiodinase in brown fat, but the physiologic significance of these effects is not clear. Type 3 5-deiodinase is found in chorionic membranes of the placenta and glial cells in the central nervous system, where it inactivates T4 by converting it to rT3 and it inactivates T3 by converting it to 3,3′-diiodothyronine (3,3′-T2) (Figure 7–14). Type 3 deiodinase levels are elevated in hyperthyroidism and decreased in hypothyroidism and, therefore, may help insulate the fetus and the brain from T4 excess or deficiency. Placental type 3 deiodinase accelerates thyroxine disposal in pregnant women, which partly explains the increased thyroxine dose requirements in treated hypothyroid women.
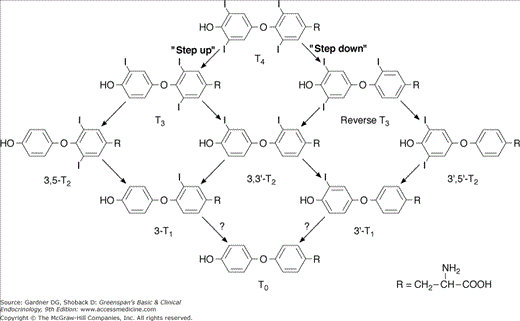
Overall, the functions of the deiodinases are physiologically important in three ways. First, they permit local tissue and cellular modulation of thyroid hormone actions. Second, they help the organism adapt to changing states, including iodine deficiency or chronic illness. Third, they regulate thyroid hormone actions in the early development of many vertebrates, including amphibia and mammals.
About 80% of T4 is metabolized by deiodination, 35% to T3 and 45% to rT3 (see Figure 7–13). The remainder is inactivated mainly by glucuronidation in the liver and biliary secretion, and to a lesser extent, by sulfation in the liver or kidney. Other metabolic reactions include deamination of the alanine side chain, forming thyroacetic acid derivatives of low biologic activity; or decarboxylation or cleavage of the ether link, forming inactive compounds.
As a result of these metabolic pathways, approximately 10% of the total 1000 nmol extrathyroidal T4 pool is cleared each day, and the plasma half-life of T4 is 7 days. Due to T3‘s lower binding affinity for plasma proteins, the turnover of its much smaller extrathyroidal pool is more rapid, with a plasma half-life of 1 day. The total body pool of rT3 is about the same size as that of T3, but rT3 has an even more rapid turnover, with a plasma half-life of only 0.2 day.
Growth and function of the thyroid gland are controlled by the hypothalamic-pituitary-thyroid axis (Figure 7–15) and, as previously discussed, by iodide through the elements of autoregulation. Hypothalamic thyrotropin-releasing hormone (TRH) stimulates thyrotrophic cells in the anterior pituitary to produce TSH, which in turn promotes thyroid gland growth and hormone secretion. In addition, deiodinases in the pituitary and peripheral tissues modulate thyroid hormone effects by their tissue-specific conversion of T4 to the more active iodothyronine T3. Finally, the molecular effects of T3 in individual tissues are modulated by the subtype of T3 receptor with which it interacts; the specific gene activation or repression response that it induces; and in a realm just now being revealed, the T3 receptor’s interaction with other ligands, closely related receptors (eg, retinoid X receptor; RXR), and coactivators and corepressors that interact with it in modulating gene expression.
Figure 7–15
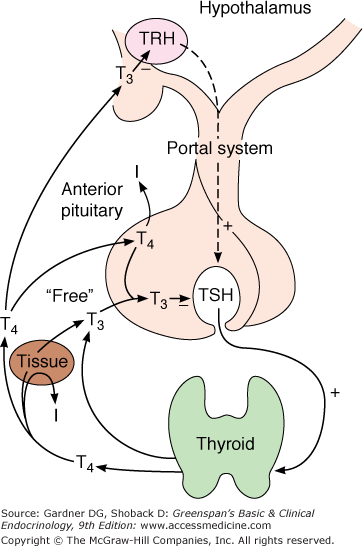
The hypothalamic-hypophysial-thyroidal axis. TRH produced in the hypothalamus reaches the thyrotrophs in the anterior pituitary by the hypothalamo-hypophysial portal system and stimulates the synthesis and release of TSH. In both the hypothalamus and the pituitary, it is primarily T3 that inhibits TRH and TSH secretion, respectively. T4 undergoes monodeiodination to T3 in neural and pituitary as well as in peripheral tissues.
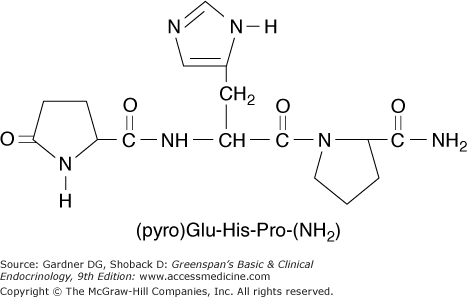
Thyrotropin-releasing hormone (TRH) is a tripeptide, pyroglutamyl-histidyl-proline amide (pyro-Glu-His-Pro-NH2), synthesized by neurons in the supraoptic and supraventricular nuclei of the hypothalamus (Figure 7–16). TRH is stored in the median eminence of the hypothalamus and then transported via the pituitary portal venous system down the pituitary stalk to the anterior pituitary, where it controls synthesis and release of TSH. TRH is also found in other portions of the hypothalamus, brain, and spinal cord, where it may have distinct functions as a neurotransmitter. The pre-pro-TRH gene, which encodes a large molecule with five copies of the TRH progenitor sequence Glu-His-Pro-Gly, is located on chromosome 3. TRH gene expression is negatively regulated by thyroid hormone—both the T3 delivered by the circulation and that arising from T4 deiodination in peptidergic neurons themselves (see Table 7–2).
In the anterior pituitary, TRH binds to a specific membrane receptor located on TSH- and prolactin-secreting cells, stimulating synthesis and release of their respective hormones. The TRH receptor is a member of the 7-transmembrane-spanning, G protein–coupled receptor family (Table 1–1; Figure 1–4). TRH binds to the receptor’s third transmembrane helix, activating both its cyclic guanosine monophosphate-producing complex and the inositol 1,4,5-triphosphate (IP3) signaling cascade that releases intracellular Ca2+ and generates 1,2-diacylglycerol, thereby activating protein kinase C. These pathways are responsible for stimulating TSH release. They coordinate transcription of the genes encoding the TSH subunits and posttranslational glycosylation of TSH, which is necessary for full biologic activity.
TRH-stimulated TSH secretion is pulsatile (Figure 7–17), with a mean TSH pulse amplitude of 0.6 mU/L every 2 hours. Normal individuals have a circadian rhythm in TSH release, with a peak circulating level between midnight and 4 AM, which is presumably controlled by a hypothalamic neuronal pulse generator driving TRH synthesis.
Figure 7–17
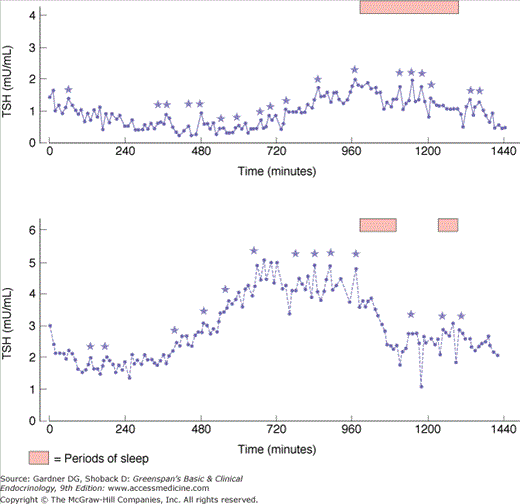
Serum TSH in two normal subjects demonstrating spontaneous pulses and the circadian rhythm of TSH secretion. (0 time is 0900; stars indicate significant pulses.)
(Reproduced, with permission, from Greenspan SL, et al. Pulsatile secretion of TSH in man. J Clin Endocrinol Metab. 1986;63:664. Copyright 1986 by The Endocrine Society.)
Thyroid hormones exert additional negative feedback on TSH production at the level of the pituitary by downregulating the number of TRH receptors on pituitary thyrotropes. Consequently, in patients with hyperthyroidism, both TSH pulses and its nocturnal surge are markedly suppressed; whereas in hypothyroid patients, the TSH pulse amplitude and nocturnal surge are much greater. In experimental animals and newborn humans, exposure to cold temperature increases TRH and TSH secretion. Certain other hormones and drugs (eg, vasopressin and α-adrenergic agonists), stimulate TRH synthesis and release.
When synthetic TRH is administered intravenously to humans as a 200 to 500 μg bolus, it generates a prompt three- to fivefold rise in the serum TSH concentration, peaking at about 30 minutes and lasting for 2 to 3 hours (see Figure 4–14). In patients with primary hypothyroidism, in whom basal TSH is elevated, there is an exaggerated TSH response to exogenous TRH; and this response is suppressed in patients with hyperthyroidism, high-dose thyroxine therapy, and central hypothyroidism.
TRH is also found in the islet cells of the pancreas, gastrointestinal tract, placenta, heart, prostate, testes, and ovaries. TRH production in these peripheral tissues is not inhibited by T3; the role of TRH in these tissues remains unknown.
Thyroid-stimulating hormone (TSH) is 28-kD glycoprotein composed of α and β subunits that are noncovalently linked. The α subunit is common to the two other pituitary glycoproteins, follicle-stimulating hormone (FSH) and luteinizing hormone (LH), and the placental hormone human chorionic gonadotropin (hCG), whereas the β subunit is unique for each glycoprotein hormone, conferring specific binding properties and biologic activity. The genes for the TSH α and β subunits are located on chromosomes 6 and 1, respectively. The human α subunit has an apoprotein core of 92 amino acids and contains two oligosaccharide chains; the TSH β subunit has an apoprotein core of 112 amino acids and contains one oligosaccharide chain. The α and β subunit amino acid chains of TSH each form three loops that are intertwined into a cystine knot (Figure 7–18). Glycosylation takes place in the RER and the Golgi, where glucose, mannose, and fucose residues and terminal sulfate or sialic acid residues are linked to the apoprotein core. These carbohydrate residues prolong its plasma half-life and enhance it ability to induce TSH receptor activation.
Figure 7–18
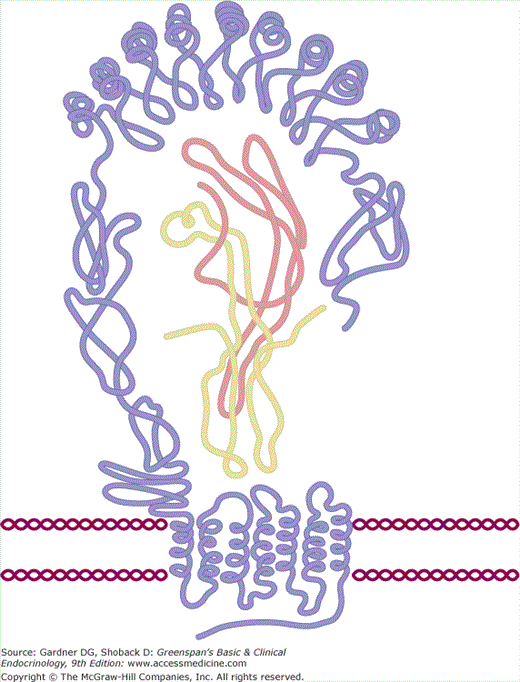
Schematic configuration of the TSH-TSHR complex. The central portion of the figure represents the ribbon-like structure of TSH within the TSH receptor. The dark blue line represents the β subunit and the light blue line the α subunit. See also Figure 7–19.
(Reproduced, with permission, from Szkudlinski MW, et al. Thyroid-stimulating hormone and thyroid-stimulating hormone receptor structure-function relationships. Physiol Rev. 2002;82:473.)
TSH controls thyroid cell growth and hormone production by binding to a specific TSH receptor, one of approximately 1000 located on the basolateral cell membrane of each thyroid cell. TSH binding activates both the cyclic adenosine monophosphate (cAMP) and the phosphoinositol pathways for signal transduction. The TSH receptor gene is located on chromosome 14; the product is a single-chain glycoprotein composed of 764 amino acids. The TSH receptor is a member of the 7-membrane spanning, G protein–coupled receptor family, with an ectodomain involved in ligand binding and an intramembrane and intracellular portion responsible for activation of the signaling pathways that promote thyroid cell growth and hormone synthesis and release (see Figure 7–19).
Figure 7–19
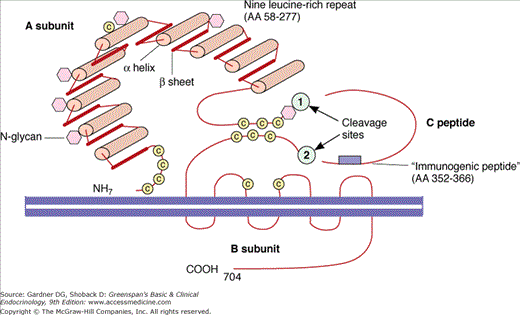
Schematic representation of the TSH receptor. The A subunit is the ligand-binding portion of the receptor and the B subunit is the activation portion. The ligands which bind to the receptor include TSH, TSH-stimulating antibody, and TSH-blocking antibody. There are two cleavage sites which allow breakage of the receptor and loss of the A subunit into the serum.
(Reproduced, with permission, from Rapoport B, et al. The thyrotropin (TSH)-receptor: interaction with TSH and autoantibodies. Endocr Rev. 1998;19:673.)
The TSH receptor is involved in the pathogenesis of numerous congenital and acquired forms of hypothyroidism and hyperthyroidism. Heritable defects resulting in impaired TSH synthesis or action have been described, including mutant genes for transcription factors required for pituitary thyrotrope differentiation (POU1F1, PROP1, LHX3, HESX1), TRH receptor, TSH β-chain, TSH receptor, and Gsα, which transduces TSH receptor binding to adenylate cyclase activation. Acquired TSH receptor blocking antibodies can also cause hypothyroidism.
The most common TSH receptor–related disorder causing hyperthyroidism is Graves disease, in which autoantibodies bind and stimulate the TSH receptor. However, the TSH receptor is involved in the etiology of several other forms of hyperthyroidism. Germline mutations activating the TSH receptor can cause familial hyperthyroidism, and somatic activating mutations result in toxic adenomas. Other mutations can lead to aberrant TSH receptor activation by hCG, the placental glycoprotein hormone that is structurally similar to TSH, in familial gestational hyperthyroidism.
TSH has many actions on the thyroid cell. Most of its actions are mediated through the G protein-adenylyl cyclase-cAMP system, but activation of the phosphatidylinositol (PIP3) system with a resulting increase in intracellular calcium is also involved. The major actions of TSH include the following:
TSH rapidly induces pseudopods at the follicular cell-colloid border, accelerating thyroglobulin resorption. Colloid content is diminished as intracellular colloid droplets are formed and lysosome formation is stimulated, increasing thyroglobulin hydrolysis and thyroid hormone release.
Individual thyroid cells increase in size; vascularity is increased; and, over a period of time, thyroid enlargement, or goiter, develops.
TSH stimulates all phases of iodide metabolism, from increased iodide uptake and transport to increased iodination of thyroglobulin and increased secretion of thyroid hormones and thyroglobulin itself. Increased NIS expression and the stimulation of cAMP production mediate increased iodide transport, and PIP3 hydrolysis and increased intracellular Ca2+ stimulate the iodination of thyroglobulin. The TSH effect on iodide transport across the cell is biphasic: initially, it is depressed (iodide efflux), and then, after a lag of several hours, iodide uptake is increased. The efflux of iodide may be due to the rapid increase in hydrolysis of thyroglobulin with release of hormone and leakage of iodide out of the gland. The TSH-mediated stimulation of thyrocyte iodine uptake and thyroglobulin secretion also occur after administration of recombinant TSH for radioiodine treatment and monitoring of patients with well-differentiated thyroid cancers.
Other effects include increased transcription of the mRNAs for thyroglobulin and TPO; increased incorporation of iodide into MIT, DIT, T3, and T4; and increased lysosomal activity, with increased secretion of T4 and T3 from the gland. There is also increased activity of type 1 5′-deiodinase, which helps conserve intrathyroidal iodine.
TSH has additional effects on the thyroid gland, including stimulation of glucose uptake, oxygen consumption, and glucose oxidation. There is accelerated turnover of phospholipids and stimulation of synthesis of purine and pyrimidine precursors, with increased synthesis of DNA and RNA.
Intact TSH and isolated α subunit are both present in circulating blood and detectable by immunoassay in concentrations that are normally 0.5 to 4.0 mU/L and 0.5 to 2 μg/L, respectively. The serum TSH level is increased in primary hypothyroidism and decreased in thyrotoxicosis, whether endogenous or from excessive oral intake of thyroid hormones. The plasma half-life of TSH is about 30 minutes, and the daily production rate is about 40 to 150 mU/d.
The glycoprotein α subunit is often disproportionately elevated in patients with TSH-secreting pituitary tumors (see later); it is also increased in normal postmenopausal women due to increased synthesis and secretion related to increased gonadotropin production.
Two major factors control synthesis and release of TSH: (1) T3 level within thyrotrophic cells, which regulates mRNA expression, TSH translation, and the hormone’s release; and (2) TRH, which controls posttranslational glycosylation and also release.
TSH synthesis and release are inhibited by high serum levels of T4 and T3 (hyperthyroidism) and stimulated by low levels of thyroid hormone (hypothyroidism). In addition, certain hormones and drugs inhibit TSH secretion. These include somatostatin, dopamine, dopamine agonists such as bromocriptine, and high doses of glucocorticoids. Severe disease may cause inhibition of TSH secretion, and there may be a rebound increase in TSH as the patient recovers. The magnitude of these effects varies; the drugs mentioned above usually only partially suppress serum TSH, but it remains detectable. In contrast, overt hyperthyroidism can suppress the TSH concentration to beneath the detection limits of even the most sensitive TSH immunoassays.
Tumors and other disorders of the hypothalamus or anterior pituitary gland can impair TRH and TSH secretion, respectively. Pituitary disorders causing hypothyroidism are termed secondary, whereas hypothalamic disease resulting in hypothyroidism is called tertiary. Differential diagnosis of these lesions is discussed later (see Thyroid Tests).
The thyroid follicle has a rich supply of capillaries that carry noradrenergic nerve fibers from the superior cervical ganglion and acetylcholine esterase-positive nerve fibers derived from the vagus nerve and thyroid ganglia. The parafollicular C cells secrete both calcitonin and calcitonin gene-related peptide (CGRP). In experimental animals, these and other neuropeptides modify thyroid blood flow and hormone secretion. In addition, growth factors such as insulin, insulin-like growth factor-1, insulin-like growth factor binding proteins, epidermal growth factor, transforming growth factor β1, and fibroblast growth factors and their receptors, as well as autocrine factors such as prostaglandins and cytokines, modify thyroid cell growth and hormone production. However, it is not yet clear how clinically important these effects are.
Pituitary type 2 5′-deiodinase converts T4 to T3 in the brain and pituitary, providing the main source of T3 within thyrotropes. Its activity increases in hypothyroidism, initially maintaining a normal intracellular T3 level in the cerebrum despite a falling plasma T4 concentration. In hyperthyroidism, a decrease in its activity moderates exposure of pituitary and neural cells to thyroid hormone excess. In contrast, type 1 5′-deiodinase is decreased in hypothyroidism, conserving T4, and increased in hyperthyroidism, accelerating T4 metabolism but also leading to increased serum levels of T3 (see Table 7–2).
Autoregulation may be defined as the capacity of the thyroid gland to modify its function to adapt to changes in the availability of iodine, independent of pituitary TSH. Thus, humans can maintain normal thyroid hormone secretion with iodide intakes varying from 50 μg to several milligrams per day. Some of the effects of iodide deficiency or excess are discussed earlier. The major adaptation to low iodide intake is the preferential synthesis of T3 rather than T4, increasing the metabolic effectiveness of the secreted hormone. Iodide excess, on the other hand, inhibits many thyroidal functions, including iodide transport, cAMP formation, hydrogen peroxide generation, and hormone synthesis and secretion. Some of these effects may be mediated by the formation of intrathyroidal iodinated fatty acids. The ability of the normal thyroid to escape from these inhibitory effects (Wolff-Chaikoff effect) allows the gland to continue to secrete hormone despite a high dietary iodide intake. It is important to note that this is different from the therapeutic effect of iodide in the treatment of Graves disease. Here, the high levels of iodide persistently inhibit thyroglobulin endocytosis and lysosomal activity, decreasing thyroid hormone release and lowering circulating hormone levels. In addition, pharmacologic amounts of iodide reduce the vascularity of the gland, which may be beneficial during thyroid surgery for Graves disease. However, this effect is also transient, lasting 10 to 14 days.
The thyroid hormones exert their actions through two general mechanisms: (1) genomic actions effected through T3 interactions with its nuclear receptors, regulating gene activity; and (2) nongenomic actions mediated by T3 and T4 interactions with certain enzymes (eg, calcium ATPase, adenylate cyclase, monomeric pyruvate kinase), glucose transporters, and mitochondrial proteins.
Thyroid hormones, that are unbound in plasma, are transported intracellularly, by either specific carriers including monocarboxylate transporter 8 (MCT8), MCT10, and organic anion transporting polypeptide (OATP1C1). OATP1C1 is expressed predominantly in brain capillaries and the choroid plexus, and transports T4 preferentially, while MCT8 and MCT10 are expressed in many tissues and transport both T4 and T3. Thyroid hormones are transported through the cell membrane into the cytoplasm, and subsequently into the nucleus, where T3 binds to its specific receptor. Clinical syndromes associated with mutations in these transporters have recently been recognized, such as the Allan-Herndon-Dudley syndrome in which affected male children develop mental retardation, myopathy, a movement disorder in association with elevated serum T3 and low serum T4 levels.
The T3 nuclear receptor is member of a nuclear receptor superfamily that includes nuclear receptors for glucocorticoids, mineralocorticoids, estrogens, progestins, vitamin D3, and retinoids (see Figure 1–16). There are two thyroid hormone receptor (TR) genes in humans: TRα located on chromosome 17 and TRβ on chromosome 3. Each gene yields at least two differentially spliced products, TRα 1 and 2 and TRβ 1 and 2, although TRα 2 is believed to be biologically inactive (see Figure 7–20). Each of these receptors has a carboxyl terminal ligand-binding domain and a centrally located DNA-binding domain with two cysteine zinc fingers that facilitate their specific attachment to thyroid hormone response elements (TREs) in the promoters of target genes and regulate their transcription (see Figures 1–16 and 1–17). The concentration of these receptors in tissue varies among tissues and with their stage of development. For example, the brain contains predominantly TRα, the liver mostly TRβ, and cardiac muscle contains both. Point mutations in the ligand-binding domain of the TRβ gene have been shown to be responsible for the syndrome of generalized resistance to thyroid hormone (GRTH). The TRs bind to TREs, which are typically paired, specific oligonucleotide sequences (eg, AGGTCA) (Figure 7–21). The TRs can also function as heterodimers with receptors for other transcription factors, such as the retinoid X and retinoic acid receptors. TREs are generally located upstream of the transcription start site for the coding regions of thyroid hormone–responsive genes. In positively regulated genes, unbound TRs interact with corepressors (eg, nuclear receptor corepressor [NCoR] and silencing mediator for retinoic and thyroid hormone receptors [SMRT]) to repress basal transcription by recruiting histone deacetylases that alter the nearby chromatin structure. When TRs are bound by T3, these corepressor complexes are released, and the T3-bound TRs associate with coactivator complexes that promote local histone acetylation; they also associate with another protein complex (vitamin D receptor-interacting protein/TR-associated proteins) that recruits RNA polymerase II and starts gene transcription. Some genes are negatively regulated by T3-bound TRs, such as pre-pro-TRH and TSH-α and -β subunit genes, but the molecular mechanisms involved are currently less well understood. Thyroid hormone’s actions to alter expression levels of specific mRNAs and their translated proteins generate a constellation of specific tissue responses (see Figure 7–21).
Figure 7–20
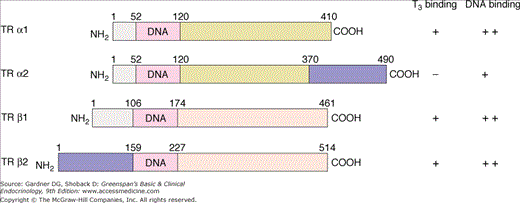
Deduced protein structure of the thyroid hormone receptor α and β gene products. The receptor protein has three domains: a DNA-binding domain with a high degree of similarity among the different types of receptors, a carboxyl terminal triiodothyronine (T3)–binding domain, and an amino terminal domain that is not required for full function. The numbers above the structures represent amino acid numbers. The properties of the receptors with respect to their ability to bind T3 and bind to a T3-response element of DNA are shown on the right. Identical shading of receptor domains indicates identical amino acid sequences. (TR, thyroid hormone receptor.)
(Reproduced, with permission, from Brent GA. The molecular basis of thyroid hormone action. N Engl J Med. 1994;331:847.)
Figure 7–21
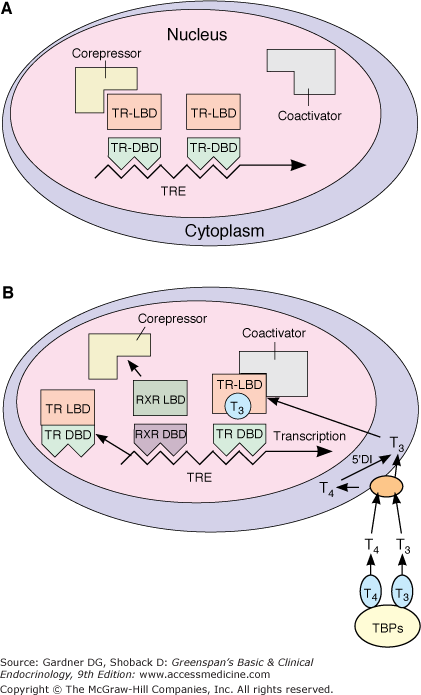
Model of the interaction of T3 with the T3 receptor. Panel A, Inactive Phase: The unliganded T3 receptor dimer bound to the TRE along with corepressors acts as a suppressor of gene transcription. Panel B, Active Phase: T3 and T4 circulate bound to thyroid-binding proteins (TBPs). The free hormones are transported into the cell by a specific transport system. Within the cytoplasm, T4 is converted to T3 by 5′-deiodinase and T3 moves into the nucleus. There it binds to the ligand-binding domain of the TR monomer. This promotes disruption of the TR homodimer and heterodimerization with RXR on the TRE, displacement of corepressors, and binding of coactivators. The TR-coactivator complex activates gene transcription, which leads to alteration in protein synthesis and cellular phenotype. (TR-LBD, T3 receptor ligand-binding domain; TRDBD, T3 receptor DNA–binding domain; RXR-LBD, retinoid X receptor ligand–binding domain; RXR-DBD, retinoid X receptor DNA–binding domain; TRE, thyroid hormone response element; TBPs, thyroxine-binding proteins; T3, triiodothyronine; T4, tetraiodothyronine, L-thyroxine; 5′DI, 5′-deiodinase.)
The nongenomic actions of thyroid hormones have long been suspected based on certain rapid tissue responses that take place before RNA transcription could occur and by recognition of T3– and T4-binding sites outside of the nucleus. For example, it has recently been shown that thyroid hormones bind the membrane protein integrin αVβ3, which is involved in thyroid hormone–mediated activation of the MAP kinase cascade and stimulation of angiogenesis.
The transcriptional effects of T3 characteristically demonstrate a lag time of hours or days to achieve full effect. These genomic actions have a number of vital effects, including tissue growth, brain maturation, increased calorigenesis and oxygen consumption, as well as other specific effects on the heart, liver, kidneys, skeletal muscle, and skin. However, some actions of T3 are believed not to be genomic, including its reduction of pituitary type 2 5′-deiodinase activity and the increased glucose and amino acid transport that it can induce in some tissues. Some specific effects of thyroid hormones are summarized below.
Iodide concentration by thyroid tissue and pituitary TSH both appear in the human fetus at about 11 weeks’ gestation. Because of the high placental content of type 3 5-deiodinase, most maternal T3 and T4 are inactivated, and very little free hormone reaches the fetal circulation. However, this small amount of free hormone from the mother may be important for early fetal brain development. After 15 to 18 weeks of gestation, the fetus is largely dependent on its own thyroidal secretion. Although some fetal growth occurs in the absence of fetal thyroid hormone secretion, brain development and skeletal maturation are markedly impaired if congenital hypothyroidism is undiagnosed and thyroid hormone therapy is not begun promptly after birth. Congenital hypothyroidism results in cretinism, the features of which include mental retardation and dwarfism.
T3 increases O2 consumption and heat production in part by stimulation of Na+-K+ ATPase in all tissues except the brain, spleen, and testis. This contributes to the increased basal metabolic rate (total somatic O2 consumption at rest) and the increased sensitivity to heat in hyperthyroidism—and the opposite in hypothyroidism. Thyroid hormones stimulate mitochondriogenesis, augmenting the cell’s oxidative capacity. They also induce changes in the mitochondrial inner membrane protein and lipid composition that increase oxidative metabolism by both genomic and nongenomic effects. The reduced efficiency of oxidative metabolism caused by thyroid hormone is also reflected in the increased futile cycling of intermediary carbohydrate metabolites.
T3 stimulates transcription of sarcoplasmic reticulum Ca2+ ATPase, increasing the rate of myocardial diastolic relaxation. It also increases expression of the more rapidly contractile isoforms of myosin heavy chain, the α isoforms, which contributes to enhanced systolic function. In myocardium, T3 also alters expression of different isoforms of the Na+-K+ ATPase genes, increases expression of α-adrenergic receptors, and decreases the concentration of the inhibitory G protein Gi α. T3 also increases the rates of both depolarization and repolarization of the sinoatrial node, increasing heart rate. Consequently, thyroid hormones have positive inotropic and chronotropic effects on the heart, which, along with the heightened adrenergic sensitivity (see later), accounts for the increased heart rate and contractility in hyperthyroidism and the reverse in hypothyroidism. Thyroid hormones also affect diastolic ventricular function, lower peripheral vascular resistance, and increase intravascular volume, which contributes further to the increase in cardiac output associated with thyroid hormone action.
Figure 7–22
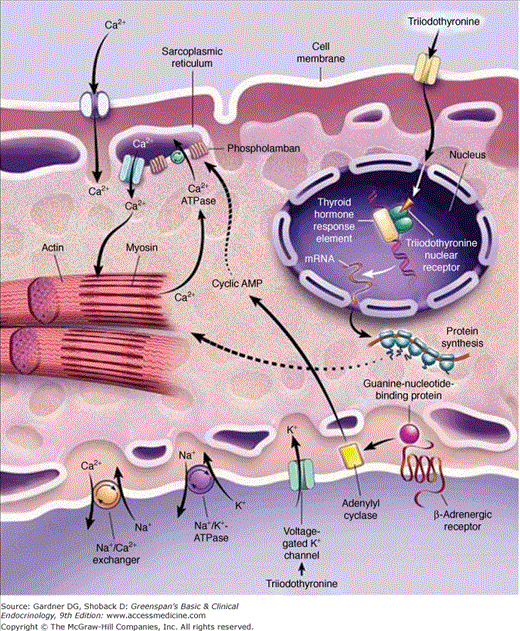
Sites of action of triiodothyronine on cardiac myocytes. Triiodothyronine enters the cell, possibly by a specific transport mechanism, and binds to nuclear triiodothyronine receptors. The complex then binds to thyroid hormone response elements of target genes and regulates transcription of these genes. Nonnuclear thyroid hormone effects on ion channels also occur.
(Reproduced, with permission, from Klein I, Ojamaa K. Thyroid hormone and the cardiovascular system. N Engl J Med. 2001;344:501.)
Thyroid hormones increase the number of β-adrenergic receptors in heart and skeletal muscle, adipose tissue, and lymphocytes. They may also amplify catecholamine action at a postreceptor site. Many of the clinical manifestations of thyrotoxicosis appear to reflect increased sensitivity to catecholamines. Furthermore, therapy with β-adrenergic blocking agents is often helpful in controlling these sympathomimetic manifestations of thyroid hormone excess.
Thyroid hormones maintain ventilatory responses to hypoxia and hypercapnia in the brain stem respiratory center. Consequently, in patients with severe hypothyroidism, hypoventilation can occur. Respiratory muscle functions are also regulated by thyroid hormone, and can be weakened in hyperthyroidism, leading to a sense of breathlessness.
The increased cellular demand for O2 in hyperthyroidism leads to increased production of erythropoietin and increased erythropoiesis. However, blood volume is usually not increased because of hemodilution and increased red cell turnover. Thyroid hormones increase the 2,3-diphosphoglycerate content of erythrocytes, allowing increased O2 dissociation from hemoglobin and increasing O2 availability to tissues. The reverse occurs in hypothyroidism.
Thyroid hormones promote gut motility, which can result in increased motility and hyperdefecation (ie, increased frequency of formed bowel movements) in hyperthyroidism. Conversely, slowed bowel transit and constipation occurs in hypothyroidism.
Thyroid hormones stimulate bone turnover, increasing bone resorption and, to a lesser degree, bone formation. Consequently, hyperthyroidism can be associated with hypercalciuria, and less often, hypercalcemia. Furthermore, chronic thyroid hormone excess can cause clinically significant bone mineral loss.
In hyperthyroidism, there is increased protein turnover and loss in skeletal muscle, which can lead to a characteristic proximal myopathy. There is also an increase in the speed of muscle contraction and relaxation, noted clinically as the hyperreflexia of hyperthyroidism and the delayed deep tendon reflex relaxation phase in hypothyroidism. A fine distal hand tremor is also typical in hyperthyroidism. As noted above, thyroid hormones are essential for normal development and function of the central nervous system, and failure of fetal thyroid function results in severe mental retardation. In the adult, hyperactivity in hyperthyroidism and sluggishness in hypothyroidism can be striking. Similarly, hyperthyroid patients can be anxious or agitated, while hypothyroidism is typically associated with a depressed mood.
Hyperthyroidism increases hepatic gluconeogenesis and glycogenolysis, as well as intestinal glucose absorption, and there may also be thyroid hormone–mediated decreases in insulin sensitivity. Thus, hyperthyroidism can worsen glycemic control in patients who also have diabetes mellitus. Cholesterol synthesis and degradation are both increased by thyroid hormones. The latter effect is due largely to an increase in hepatic low-density lipoprotein (LDL) receptor number, accelerating LDL clearance. Consequently, total and LDL cholesterol levels are typically elevated in patients with hypothyroidism. Lipolysis is also increased, releasing fatty acids and glycerol into circulating plasma.
Thyroid hormones alter the production, responsiveness, and metabolic clearance of a number of hormones. In hypothyroid children, impaired growth hormone release slows longitudinal growth. Hypothyroidism can cause delayed puberty by impairing gonadotropin-releasing hormone (GnRH) and gonadotropin secretion. Conversely, primary hypothyroidism can also cause precocious puberty, perhaps as an effect of very high TSH levels on gonadotropin receptors. In adults, hypothyroidism causes hyperprolactinemia in a minority of affected women. Menorrhagia (prolonged and heavier menses) and anovulation are common in hypothyroid women, the latter resulting in infertility. The responsiveness of the hypothalamic-pituitary-adrenal axis to stress is blunted in hypothyroid patients. A slowing of the cortisol metabolic clearance rate compensates for this in the hypothyroid state. Conversely, however, restoration of euthyroidism can rarely provoke adrenal insufficiency as cortisol metabolism is accelerated in patients with diminished cortisol reserve due to concomitant disease affecting the adrenal axis.
In hyperthyroidism, accelerated aromatization of androgens to estrogens and increased sex hormone–binding globulin levels contribute to the gynecomastia and elevated total testosterone levels seen in affected men. Hyperthyroidism can also impair normal GnRH and gonadotropin regulation of ovulation and menses, causing infertility and amenorrhea, respectively. All of the endocrine derangements occurring with thyroid dysfunction are reversed by appropriate treatment to restore euthyroidism.
Before the fetus develops its own independent thyroid function, it depends on maternal thyroid hormones for early neural development. By the 11th week of gestation, however, the hypophysial portal system has developed, and measurable TSH and TRH are present. At about the same time, the fetal thyroid begins to trap iodine. The secretion of thyroid hormone probably begins in mid gestation (18-20 weeks). TSH increases rapidly to peak levels at 24 to 28 weeks, and T4 levels peak at 35 to 40 weeks. T3 levels remain low during gestation; T4 is converted to rT3 by type 3 5′-deiodinase during fetal development. At birth, there is a sudden marked rise in TSH stimulated by exposure to the colder extrauterine environment, with a rise in T4, a rise in T3, and a fall in rT3. These parameters gradually stabilize over the first month of life.
A number of thyroid parameters change during pregnancy. There is an increase in urinary iodide clearance; in areas of low dietary iodine intake, this can cause maternal goiter, or when severe, hypothyroidism. During pregnancy, there is a rise in TBG due to an estrogen-induced increase in hepatic glycosylation of TBG, prolonging its metabolic clearance rate. Consequently, serum total T4, and to a lesser extent, total T3 concentrations rise. Rising levels of hCG, which has weak TSH receptor agonist activity, contributes to minimal thyroid enlargement. Maternal hCG levels peak at approximately 12 weeks resulting in a transient high-normal or even modestly elevated serum-free T4 level and a physiologic suppression of serum TSH to the low-normal or even subnormal range. Pathologically elevated hCG levels, found in women with a molar pregnancy or choriocarcinoma, can cause overt hyperthyroidism. Women with severe vomiting in pregnancy, called hyperemesis gravidarum, may also have higher levels of hCG, and can develop a transient gestational thyrotoxicosis. As noted earlier, placental type 3 deiodinase accelerates thyroxine clearance during pregnancy, which partly accounts for the increased thyroxine dose requirement in treated hypothyroid women, and occasionally causes mild hypothyroidism in women with autoimmune thyroiditis and decreased thyroid gland reserve.
Transplacental delivery of thyroid-related substances can have consequences for the fetus. Maternal iodide crossing the placenta is essential for fetal thyroid hormone production; however, large amounts of iodide can actually inhibit fetal thyroid function and cause a goiter substantial enough to obstruct delivery. In women with autoimmune thyroid disease, maternal immunoglobulins that stimulate or block the TSH receptor can cross the placenta and cause thyroid dysfunction in the fetus. The antithyroid drugs methimazole and PTU cross the placenta, and in large doses will impair fetal thyroid function (see Chapter 16).
T4 turnover is highest in infants and children, and gradually falls to adult levels after puberty. The T4 turnover rate is then stable until after age 60, when it again begins to decline. In the elderly, the metabolic clearance of T4 can decrease by as much as 50%, necessitating a reduction in T4 dose for treated patients.
Acute or chronic illness has several striking effects on thyroid function and hormone economy. The most common and earliest effect is inhibition of T4-to-T3 conversion, with a resulting decrease in the circulating T3 level. This change is accompanied by a rise in the serum reverse T3 level, due to both its decreased conversion to 3,3′-T2 and some acceleration of its production from T4. These changes occur physiologically in the fetus and pathologically in circumstances of caloric restriction, as in malnutrition, starvation, and anorexia nervosa, and in patients with a variety of acute or chronic systemic illnesses (Table 7–3). The pathogenesis of this low T3 syndrome is thought to involve cytokines, such as tumor necrosis factor, secreted by inflammatory cells, which inhibit type 1 5′-deiodinase. Drugs can also inhibit type 1 5′-deiodinase and lower the serum T3 levels in treated patients; these agents include corticosteroids, amiodarone, iodinated cholecystographic dyes, PTU, and high-dose propranolol. Initially, low serum T3 levels are typically accompanied by total and free T4 levels that are normal or, less commonly, slightly elevated (Figure 7–23). As illness becomes more severe, however, there is a decline in serum total and free T4 as well. This hypothyroxinemia can have several causes. In certain illnesses (eg, nephrosis and severe liver disease), there can be a decline in the circulating TBG concentration. A rise in plasma free fatty acids can interfere with T4 binding to TBG, liberating more free T4 that, in turn, feeds back on the hypothalamus and pituitary to reduce TSH and establish a new equilibrium with a lower total T4. TSH can also be suppressed by cytokines circulating in severely ill patients and by the dopamine or corticosteroids often used to treat critically ill patients. Finally, T4 clearance is accelerated in illness. All of these factors can contribute to the lower serum levels of T4 and free T4, as measured by free hormone immunoassays and estimated T3 resin-uptake procedures. However, the free T4 level measured by equilibrium dialysis has been reported to be normal in most, but not all patients. This suggests that the low serum free T4 levels reported by most commercially available free T4 assays may be a measurement artifact that is unique to critically ill patients.
Figure 7–23
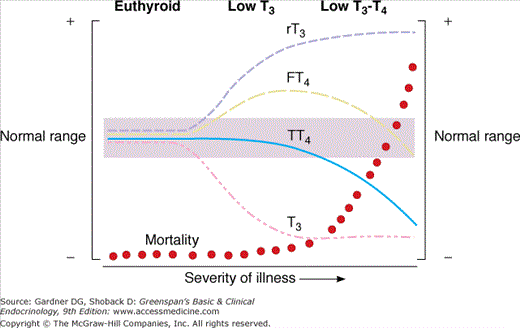
A schematic representation of the changes in serum thyroid hormone values with increasing severity of nonthyroidal illness. A rapidly rising mortality rate accompanies the fall in serum total T4 (TT4) and free T4 (FT4) values.
(Reproduced, with permission, from Nicoloff JT. Abnormal measurements in nonendocrine illness. In: Medicine for the Practicing Physician. 2nd ed. Hurst JW, ed. Butterworth-Heinemann; 1991.)
1. Fetal life 2. Caloric restriction 3. Hepatic disease 4. Major systemic illness 5. Drugs:
6. Selenium deficiency |
Two clinical questions arise in sick patients with this constellation of thyroid function test abnormalities. First, it can be challenging to distinguish the euthyroid sick syndrome from central hypothyroidism; in both conditions, the T4 and TSH levels can be low. Clinical information can be helpful, including a history of known pituitary or thyroid disease, head trauma or surgery, or manifestations of other elements of hypopituitarism (eg, small testes in a man). Additional laboratory testing can sometimes be informative. The serum free T4 level determined by equilibrium dialysis is typically normal in the euthyroid sick syndrome. In postmenopausal women, an elevated serum FSH attests to at least one intact pituitary function, although gonadotropin levels can be low in severe illness as well. Measurement of serum cortisol may be particularly helpful, because it should be normal or elevated in any patient with significant systemic illness. If it is normal or elevated, hypothalamic or pituitary disease is most unlikely. Sometimes, however, it is necessary to obtain emergent imaging of the hypothalamus or pituitary to resolve the question. These thyroid function test abnormalities normalize when the patient recovers. However, recovery can be accompanied by transient rebound elevation of the serum TSH that may be misinterpreted as hypothyroidism. In this setting, in the absence of clinically apparent hypothyroidism, it is best to avoid thyroid hormone therapy and to reevaluate a few weeks following recovery.
The second clinical question is whether the euthyroid sick syndrome itself should be treated with thyroid hormone. Certainly, many critically ill patients have clinical problems that are reminiscent of hypothyroidism, such as hypothermia, altered sensorium, ventilatory and myocardial dysfunction, and hyponatremia. It has been postulated that intracellular hypothyroidism may exist in these patients and that thyroid hormone therapy might be beneficial. Conversely, it has been proposed that these changes in thyroid status are, in fact, compensatory mechanisms for reducing rates of oxygen consumption and protein catabolism and that thyroid hormone therapy might actually be harmful. Furthermore, two randomized trials, small clinical series, and animal models of systemic illness all have failed to demonstrate any benefit of thyroid hormone therapy in this setting. Although the issue remains unresolved, current practice is generally to withhold thyroid hormone therapy unless there is convincing clinical and biochemical evidence of true hypothyroidism.
Autoimmunity is involved in the pathogenesis of many thyroid diseases, including hyperthyroid Graves disease, hypothyroidism with autoimmune or Hashimoto thyroiditis, silent and postpartum thyroiditis, and certain forms of neonatal thyroid dysfunction. Immunologic defense against foreign substances and neoplastic cells involves macrophages that ingest and digest the foreign material and present peptide fragments on the cell surface in association with a class II protein encoded by the HLA-DR region of the MHC gene complex. This complex is recognized by a T-cell receptor on a CD4 helper T cell, which stimulates the release of cytokines such as interleukin-2 (IL-2). These cytokines amplify the response by inducing T-cell activation and proliferation, induction of killer cell activity in CD8 suppressor cells, and stimulation of antibody formation against the foreign antigen by B lymphocytes. Eventually, the activation process is muted by the action of the CD8 suppressor cells (see Chapter 2).
Early observations suggested the possibility that immunologic reactions could affect the thyroid gland. Thyroiditis was induced by immunization of mice with thyroid tissue and Freund adjuvant. Thyroglobulin antibodies were identified in the sera from patients with thyroiditis, and a long-acting thyroid stimulator, later shown to be an antibody to the TSH receptor, was found in the sera of patients with Graves disease.
There are three major thyroidal autoantigens: thyroglobulin, TPO, and the TSH receptor. Circulating autoantibodies to these antigens are useful markers for thyroid autoimmunity, but T-cell-mediated immune mechanisms are central to the pathogenesis of thyroid diseases. Thyroid cells have the capacity to ingest antigen (eg, thyroglobulin) and, when stimulated by cytokines such as gamma interferon, express cell surface class II molecules (eg, HLA-DR4) that present these antigens to T lymphocytes. In addition to the antigen and the class II molecule, other factors such as costimulatory signals from antigen-presenting cells, are doubtless required to initiate autoimmune thyroid disease. One hypothesis is that the process is initiated or promoted by external antigens (eg, infectious agents) that lead to antibody and cellular immune responses through cross-reactivity. In addition, genetic factors clearly predispose individuals to thyroid autoimmunity. HLA typing in patients with Graves disease reveals a high frequency of HLA-B8 and HLA-DR3 in Caucasians, HLA-Bw46 and HLA-B5 in Chinese, and HLA-B17 in blacks. In Caucasians, atrophic thyroiditis has been associated with HLA-B8 and goitrous Hashimoto thyroiditis with HLA-DR5. It has been suggested that a genetically induced antigen-specific defect in suppressor T lymphocytes may be the basis for autoimmune thyroid disease.
The increased incidence of autoimmune thyroid disease in postpubertal and premenopausal women, as well as the occurrence of postpartum thyroiditis, implies a role for female sex hormones in the pathogenesis of autoimmune thyroid disease. A high iodine intake may result in more highly iodinated thyroglobulin that is more immunogenic and would favor the development of autoimmune thyroid disease. Therapeutic doses of lithium, used for the treatment of bipolar disorder, can interfere with suppressor cell function and may precipitate autoimmune thyroid disease.
1In this chapter, the words “iodine,” referring to the uncharged element, and “iodide,” referring to its negatively charged anion form, are used interchangeably.
2Jod is German for iodine; Carl Adolph von Basedow was one of the first physicians to describe hyperthyroidism.
Tests of Thyroid Function
The high prevalence of thyroid dysfunction and neoplasia make testing for thyroid disease a daily necessity in virtually every field of medical practice—whether a pediatrician screening a neonate for congenital hypothyroidism, an internist considering possible thyroid dysfunction in a patient with nonspecific complaints, an obstetrician-gynecologist evaluating a woman with infertility, a surgeon assessing a thyroid nodule, or virtually any type of specialist considering thyroid disease as the potential cause for a complaint or test abnormality in the field of interest (eg, cardiologist, gastroenterologist, neurologist, psychiatrist, ophthalmologist).
Several levels of laboratory and radiologic investigation for thyroid diseases should be considered. First, it is common for clinicians with a relatively low index of suspicion for thyroid dysfunction in a particular patient to want to exclude its presence with greater certainty. For this purpose, a test with high sensitivity (ie, few false-negative results), wide availability, and low cost is most appropriate. The serum TSH concentration often plays this role in clinical consideration of hypothyroidism and hyperthyroidism. Second, when suspicion of thyroid disease is moderate to high, based on clinical or other routine laboratory results, testing with even greater sensitivity to detect rare thyroid disorders is required, as is a test that can begin to define the severity and underlying cause of thyroid dysfunction. For example, in a patient with hypothermia, confusion, periorbital edema, and hypercholesterolemia, measurement of both the serum TSH and free T4 concentrations would permit exclusion of both primary and central forms of hypothyroidism; furthermore, if hypothyroidism is confirmed, the site of responsible disease would be implicated. Third, when thyroid dysfunction is clinically obvious or already biochemically confirmed by first-line testing, then more sophisticated studies are sometimes indicated to identify the underlying cause and plan appropriate therapy. For example, in a postpartum woman with thyrotoxicosis, an additional serum T3, thyroid-stimulating immunoglobulin titer, and radionuclide thyroid uptake may be needed to differentiate Graves disease and postpartum thyroiditis—for which there are distinct therapeutic approaches.
Because thyroid dysfunction usually arises from primary disorders of the thyroid gland, serum TSH measurement is the most widely employed test to determine whether thyroid dysfunction exists. The sensitivity of the hypothalamic-pituitary-thyroid axis ensures that both primary hypothyroidism and thyrotoxicosis, due to either primary thyroid disorders or exogenous thyroid hormones, are detected. Furthermore, even mild degrees of thyroid dysfunction, in which the serum thyroid hormone concentrations have fallen or risen significantly but remain within the broad reference range (ie, subclinical or mild thyroid dysfunction) are identified. The inverse logarithmic relationship between serum TSH and free T4 concentration in various situations is shown in Figures 7–24A and 7–24B. TSH assays must be sensitive (ie, able to distinguish low from very low and undetectable TSH levels) to be used for diagnosis of thyrotoxicosis. A National Academy of Clinical Biochemistry guideline specifies that sensitivity, or lower limit of detection, for TSH assays should be less than 0.02 mU/L. It is better to assess and compare TSH assays by this quantitative criterion rather than marketing terms such as third generation or ultrasensitive.
Figure 7–24
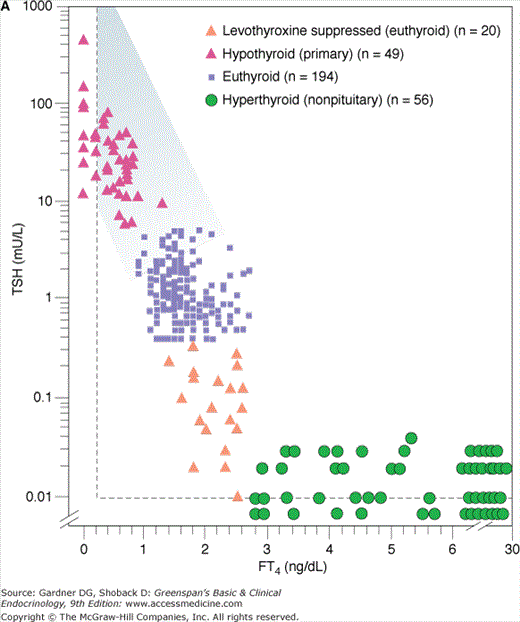
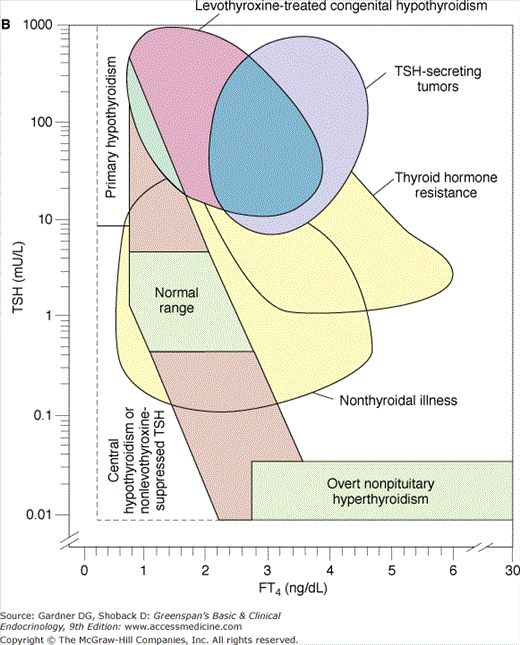
A. Relationship between serum-free thyroxine by dialysis (FT4) ng/dL and log10 TSH in euthyroid, hyperthyroid, hypothyroid, and T4-suppressed euthyroid individuals. Note that for each unit change in T4 there is a logarithmic change in TSH. B. Relationship between FT4 by dialysis and log10 TSH in normal subjects and subjects with various illnesses including primary hypothyroidism, central hypothyroidism and non–levothyroxine-suppressed TSH, levothyroxine-treated congenital hypothyroidism, TSH-secreting tumors, thyroid hormone resistance, nonthyroidal illness, and nonpituitary hyperthyroidism.
(Reproduced, with permission, from Kaptein EM. Clinical applications of free thyroxine determinations. Clin Lab Med. 1993;13:654.)
There are, in general, two sorts of immunoassays used to measure TSH (and T4 and T3) in serum samples: immunometric assays (IMA) and radioimmunoassay (RIA). In an IMA or sandwich assay, an antibody—usually a mouse monoclonal antibody—directed against one epitope on the TSH molecule is fixed to a solid matrix, and a second monoclonal TSH antibody that binds another TSH epitope is labeled with a detectable marker, which can be a radioisotope, a colorimetrically quantifiable enzyme, or a fluorescent or chemiluminescent tag. In such IMAs, the concentration of TSH, which links the solid state and labeled antibodies, is proportionate to the intensity of signal emitted by the marker once unbound second antibody has been separated off. In contrast, in conventional RIAs, a small amount of a TSH tracer, to which a radioactive molecule has been linked, competes for binding to first antibody (eg, a rabbit antihuman TSH polyclonal antibody). Then antibody-bound TSH, both in the sample and the tracer, is separated from the free tracer in the supernatant using one of several techniques: a second antibody directed against the first (eg, goat antirabbit immunoglobulin antibody), polyethylene glycol, or staphylococcal protein A. In RIAs, the concentration of TSH in the sample is inversely proportionate to tracer activity. In general, TSH RIAs are less sensitive and less widely employed than IMAs.
There are infrequent but important limitations of the serum TSH assay alone for diagnosis of thyroid dysfunction when clinical suspicion of hypothyroidism is high and in certain other special circumstances (Table 7–4). First, central hypothyroidism can be overlooked. In patients with hypothyroidism due to a pituitary tumor, for example, the serum TSH is typically not elevated, but rather low or inappropriately normal. Consequently, simultaneous measurements of both the serum free T4 and TSH would be required to further substantiate or exclude this diagnosis. Central hypothyroidism can also be confirmed by assessing the serum TSH response to an injection of TRH. To perform this test, serum samples for TSH are obtained before and 30 to 60 minutes after 200 μg of TRH is administered intravenously. The absence of a significant rise in serum TSH concentration (eg, to a peak value >7 mU/L) indicates pituitary or hypothalamic disease or suppression by exogenous thyroid hormone. A modest and delayed rise may be seen in patients with hypothalamic disease and tertiary hypothyroidism. Currently, TRH is not available in the United States. Another cause of a misleadingly low serum TSH concentration is drug treatment with either high-dose corticosteroids or dopamine, both of which inhibit TSH secretion (see Table 7–4).
Clinical hyperthyroidism TSH-secreting pituitary adenoma Isolated pituitary resistance to thyroid hormone |
Clinical hypothyroidism Central (pituitary or hypothalamic) hypothyroidism Preceding TSH suppression (eg, recently treated hyperthyroidism) |
Clinical euthyroidism Systemic illnesses (↓ during acute phase, ↑during recovery) Generalized resistance to thyroid hormone (compensated) Assay interference
Drugs: dopamine, dobutamine, glucocorticoids |
A second limitation of TSH testing alone is that there are other nonthyroidal causes of an elevated serum TSH concentration, as described in Table 7–4.
The concentrations of total and free (unbound) T4 and T3 are measurable by a variety of automated assay techniques. Serum total thyroid hormone concentrations are widely available and accurate for assessment of most patients with overt thyroid dysfunction. However, a limitation of these assays is they can provide false-positive and false-negative results when changes in the concentration or binding affinity of the thyroid hormone–binding plasma proteins are altered. For example, a pregnant woman with an estrogen-induced increase in the concentration of TBG could be misdiagnosed as being hyperthyroid if only a total T4 assay was employed. Conversely, a pregnant patient with primary hypothyroidism might be overlooked if the elevated TBG concentration produced total T4 within the reference range. Despite these reservations, diagnostic accuracy is high when total T4 assay results are interpreted along with TSH assay findings.
These problems are circumvented by measuring the free thyroxine (FT4) concentration by free T4 IMAs or equilibrium dialysis or estimating it using the free thyroxine index (FT4I), also called the thyroid hormone–binding ratio (THBR). Free T4 assays employ strategies to measure only the unbound fraction of T4 in serum samples. One approach is to employ a competing thyroxine analog tracer that binds to the assay antibody but not to plasma proteins. Such free T4 assays are quite accurate in distinguishing straightforward TBG derangements from true FT4 abnormalities. However, they may be influenced to some extent by variations in total T4
Related posts:
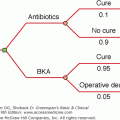


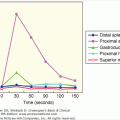
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


