Fig. 6.1
Regulation of placental inflammatory response by dietary fatty acids. Saturated and monounsaturated fatty acids activate TLR4 leading to downstream inflammatory pathways JNK and NF-κB. JNK activates AP-1 transcription factors, while NF-κB subunits directly translocate to the nucleus and bind promoter regions of proinflammatory cytokines. Omega-3 fatty acids bind to the GPR120 plasma membrane receptor to inhibit inflammatory pathways. The intracellular NLRP3 inflammasome, which is responsible for IL-1β maturation, is also activated by dietary fatty acids and inhibited by omega-3 fatty acids. AP-1 activating protein-1, GPR120 G protein-coupled receptor 120, IKK-β inhibitor of nuclear factor kappa B kinase, JNK c-jun-N-terminal kinase, MUFA monounsaturated fatty acid, NF-κB nuclear factor kappa B, NLRP3/Inflammasome nod-like receptor 3 inflammasome, SFA saturated fatty acid
6.3 Placental Inflammation in Maternal Obesity
While there have been numerous animal models of maternal obesity in pregnancy demonstrating placental inflammation [18, 32–34], we have limited our discussion to human studies. Several studies show increased expression of proinflammatory cytokines IL-6, IL-1β, and TNFα in placental tissues of obese mothers [8, 10–12]. The mechanism(s) of placental inflammation in maternal obesity, however, has been a source of contention. Analogous to adipose tissues, increased accumulation of maternal macrophages in placentas of obese mothers has been reported by some investigators [9, 10], while others were unable to show any differences [11]. Similarly, Saben et al. have implicated placental JNK and NF-κB with maternal obesity [12, 35], while we could not find any changes in these pathways, but discovered increased STAT3 and p38 MAPK activity in placentas of obese mothers [8]. One possible explanation for these differences may be related to subject selection, since maternal obesity is also associated with several comorbidities including hypertension and gestational diabetes, conditions potentially more likely to invoke a greater multisystemic inflammatory response than obesity per se.
While activation of placental inflammatory processes in maternal obesity has been described [8, 12, 35, 36], the physiological significance of placental inflammation has not been addressed in adequate detail. Inflammation of the fetal membranes (chorioamnionitis) is a major risk factor for preterm birth, which may result in premature rupture of the membranes (PROM). Recent epidemiological studies demonstrate increased risk of preterm deliveries in obese mothers [37, 38]. However, it is currently unclear if placental inflammation associated with maternal obesity increases the risk of PROM. Systemic or placental inflammation may also result in endothelial dysfunction. Pre-gravid obesity increases maternal levels of markers of endothelial dysfunction [39] and is associated with impaired vascular function in placental arteries [17]. Consequently, this may impact upon fetal heart development, because uteroplacental circulation has been linked with fetal vascular function [40].
Previous work from our lab demonstrated that proinflammatory cytokines influence placental nutrient transport functions [41, 42]. Using concentrations similar to circulating cytokine concentrations in obese women, IL-6 and TNFα were shown to stimulate amino acid transport activity [41], while higher concentrations were required to stimulate fatty acid uptake into primary trophoblasts [42]. STAT3 activity was required for IL-6 (and leptin)-stimulated amino acid uptake [41, 43], while our preliminary data suggests a role for p38 MAPK in regulating the TNFα response (Aye et al, 2015 unpublished observations). In addition to cytokines, insulin and leptin (hormones elevated in maternal obesity) also significantly increase placental amino acid transport [44, 45]. Interestingly, IL-1β which is also upregulated in the placentas of obese mothers [8, 10, 11] was shown to inhibit insulin-dependent [46] and insulin-independent [47, 48] amino acid transport in placental primary trophoblasts and trophoblast cell lines. These findings suggest complex interactions between proinflammatory cytokines and maternal hormones in the regulation of placental nutrient transfer to the fetus.
6.4 The Emerging Role of Placental Inflammasomes in Pregnancy Disorders
Inflammasomes are multi-protein complexes composed of a cytoplasmic receptor, an apoptosis-associated speck-like protein (ASC) containing a caspase activation and recruitment domain (CARD) adaptor, and pro-caspase-1, which associate upon cellular exposure to microbial or endogenous danger signals. Upon activation, the inflammasome complex formation results in caspase-1 maturation leading to proteolytic cleavage of pro-IL-1β into mature IL-1β (Fig. 6.2). This is then followed by either secretion of IL-1β into the circulation which may lead to systemic inflammatory effects or more locally induced pyroptosis. Currently, six members of the nod-like receptor (NLR) family are known to function as “pathogen” or “danger” sensors to initiate inflammasome complex formation. Of these, NLRP3 is best characterized for its role in both immune and metabolic disorders.
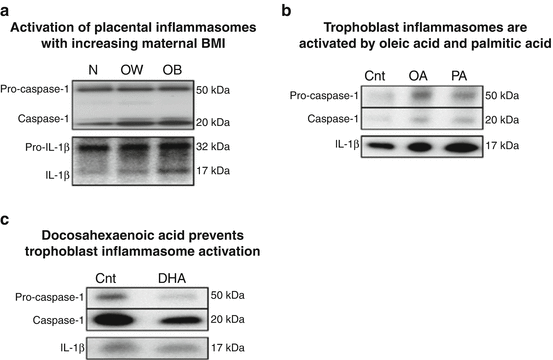
Fig. 6.2
Increased placental inflammasomes in maternal obesity – regulation by dietary fatty acids. (a) Representative immunoblot showing caspase-1 activation and increased IL-1β maturation with maternal adiposity. (b) Immunoblot demonstrating increased inflammasome activation by oleic and palmitic acid (100 μM) in primary human trophoblasts. (c) Immunoblot of decreased inflammasome activation by 50 μM DHA treatment in primary human trophoblasts. BMI body mass index, DHA docosahexaenoic acid, N normal, OW overweight, OB obese, Cnt BSA-control, OA oleic acid, PA palmitic acid
The placenta expresses the cytosolic NLRP1, NLRP3, and NLRC4 [49], which respond to multiple factors associated with both pathogenic and sterile inflammation. Activation of inflammasomes in gestational tissues has been reported in a number of pregnancy complications including preterm birth [50–52], preeclampsia [53–55], and microbial infections [56]. The resulting release of mature IL-1β mediates a myriad of effects in gestational tissues including induction of IGFBP-1 expression in decidual tissues thereby decreasing the bioavailability of IGF-I in the feto-maternal interface and in the maternal circulation [57] and stimulating placental production of progesterone [58], hCG [59], and activin-A [60]. Furthermore, IL-1β is a highly apoptotic agent in human gestational tissues [61]. Hence knowledge of the mechanisms governing IL-1β production in the placenta plays a critical role in understanding the pathogenesis of several pregnancy disorders.
We recently identified increased caspase-1 activation in placentas of women with high BMI ([62] and Fig. 6.2), despite no changes in placental NF-κB DNA-binding activity. Likewise, expression of the mature IL-1β protein (17 kDa) in placental tissues was also positively correlated with maternal BMI, whereas pro-IL-1β (32 kDa) was not significantly altered by maternal BMI. These findings indicate a novel mechanism in which IL-1β is regulated at the posttranslational level by maternal obesity. We further determined the impact of IL-1β on placental function and demonstrated that IL-1β inhibits insulin signaling and function (as measured by insulin-mediated amino acid transport) in primary human trophoblasts [46]. Inhibition of insulin signaling was mediated at the level of the insulin receptor substrate-1 (IRS-1), where IL-1β increased the inhibitory serine phosphorylation of IRS-1 (Ser307), decreased tyrosine phosphorylation-mediated activation of IRS-1 (Tyr612), and reduced total IRS-1 levels. Decreased IRS-1 activity led to inactivation of both the PI3K and GRB2 signaling pathways downstream of IRS-1. Taken together, these findings suggest that the increased inflammasome activation in placentas of high BMI mothers may lead to decreased placental insulin sensitivity. However, the signals activating inflammasomes in these placentas are currently unclear.
Because the classical inflammatory pathways associated with the microbial response (namely, NF-κB and JNK) were not altered in the placenta in pregnancies complicated by maternal obesity, we hypothesized that placental inflammasome activation is a result of DAMPs or nutritional and/or oxidative stress. Robust activation of the inflammasomes requires two signals: a first “priming” signal, which promotes NF-κB activity leading to pro-IL-1β expression, and a second “activating” signal that activates caspase-1. In the placenta, constitutive NF-κB activity is likely to contribute to pro-IL-1β expression, whereas caspase-1 activation requires a specific event. DAMPs including cholesterol crystals, ATP, potassium efflux, and ceramide initiate caspase-1 maturation in many cell types [63]. However, it is currently unclear if DAMPs provide the mechanistic link to placental inflammasome activation because these factors have not been implicated in maternal obesity. On the other hand, maternal obesity is associated with oxidative stress, which may provide the necessary signal required for caspase-1 activation [12, 14, 64]. Dietary SFAs activate inflammasomes in a cell-specific manner [65, 66]. In immune cells palmitic acid, but not oleic acid, triggered inflammasome activation [67]. In contrast, both these saturated fatty acids increased caspase-1 activity and IL-1β maturation in cultured primary human trophoblast cells (Fig. 6.2). This discrepancy is particularly relevant in pregnancy because obese mothers typically have high circulating levels of SFA, especially palmitic acid and monounsaturated fatty acid (MUFA) such as oleic acid [68]. Interestingly, both palmitic and oleic acids are capable of activating TLR4 [35, 69], suggesting that these fatty acids stimulate both the necessary pathways required for robust inflammasome activation. Moreover, most Western diets have low levels of long-chain n-3 PUFA – in particular docosahexaenoic acid (DHA) [70], which has previously been shown to prevent inflammasome activation in macrophages [71–73]. Consistent with these reports, our preliminary data demonstrates that DHA decreases (pro-)caspase-1 expression thereby attenuating IL-1β maturation (Fig. 6.2). The high dietary SFA and MUFA combined with low n-3 PUFA may therefore favor inflammasome activation in the placentas of high BMI mothers (Fig. 6.3).
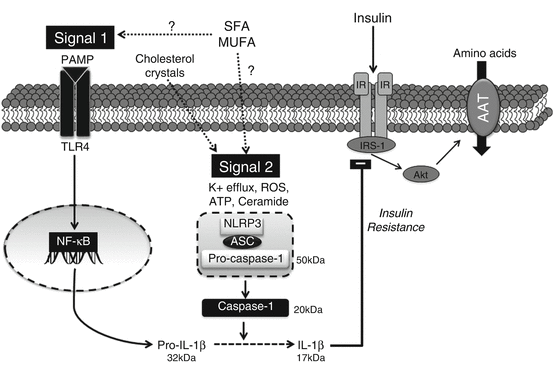
Fig. 6.3
Mechanisms linking dietary inflammasome activation to placental insulin resistance. Inflammasome-mediated IL-1β maturation requires a “priming” signal involving TLR4-NF-κB-mediated pro-IL-1β expression. A secondary “activation” signal in the form of DAMPs or dietary fatty acids promotes NLRP3-inflammasome complex formation resulting in caspase-1-mediated proteolytic cleavage of IL-1β. Mature IL-1β may function in an autocrine manner to degrade IRS-1 protein leading to decreased insulin signaling and insulin-mediated amino acid transport. AAT amino acid transport, ASC apoptosis-associated speck-like protein containing a CARD adaptor, IR insulin receptor, IRS-1 insulin receptor substrate-1, MUFA monounsaturated fatty acid, NF-κB nuclear factor kappa B, NLRP3 nod-like receptor 3, PAMP pathogen-associated molecular pattern, ROS reactive oxygen species, SFA saturated fatty acid, TLR4 toll-like receptor 4
The clinical significance of increased inflammasome activation leading to IL-1β-mediated insulin resistance in the placenta may be substantial. Although, increased placental amino acid transport activity has been reported in obese [13] or diabetic mothers [74], other studies demonstrate either no changes [75] or reduced transport activity [76, 77]. The discrepancy between these reports may be due to the inflammatory status of these placentas. It is possible that an increased placental inflammatory response associated with maternal obesity [8, 10–12, 35] or diabetes [78–81] may limit placental insulin-mediated transport, featuring an adaptive mechanism to limit excessive fetal growth in the presence of maternal hyperinsulinemia. Providing circumstantial support for this concept are reports demonstrating that the inflammatory processes associated with fetal growth restriction also inhibit placental transport activity in pregnancies complicated by malaria, a condition which increases the risk of fetal growth restriction [48].
Glyburide is a sulfonylurea agent which stimulates insulin release in the pancreas. The use of glyburide in the management of gestational diabetes has been actively explored, due to its glucose-lowering effects. While generally considered a safe alternative to insulin treatment in gestational diabetes pregnancies [82], a recent meta-analysis demonstrated that glyburide use in pregnancy is associated with increased fetal growth [83]. Although it is currently unclear if the association represents a cause and effect, it is interesting to note that glyburide is a potent inhibitor of inflammasome activation [84]. Hence inhibition of inflammasomes by glyburide may reduce IL-1β maturation, resulting in unrestricted insulin signaling in the placenta. Because insulin stimulates placental amino acid transport [45, 85], this may result in increased nutrient transfer to the fetus. In vitro studies testing this hypothesis will establish a mechanistic link between glyburide use in pregnancy and fetal growth.
6.5 Conclusions
The incidence of maternal obesity and its comorbidities (diabetes and cardiovascular disease) continues to surge, with major public health ramifications. Maternal obesity not only affects newborn health, but it also impacts the long-term health of the child leading to increased risk of childhood obesity and diabetes. Given all the evidence for the critical role of the in utero environment for lifelong health, understanding the impact of obesity in pregnancy represents a significant challenge, but also an intriguing opportunity to improve the health of future generations. Obesity is associated with the triad of metabolic complications: insulin resistance, dyslipidemia, and inflammation. Studies in recent years have demonstrated intimate links between these metabolic pathways, for example, inflammation causes insulin resistance and dyslipidemia, and dyslipidemia causes inflammation and insulin resistance. Excess nutrients, in particular dietary fatty acids, have emerged as a common instigator of these metabolic complications influencing insulin signaling and inflammatory pathways. As an organ of exchange, the placenta is a critical mediator of fetal health and is highly responsive to maternal health and diet. We recently reported inflammasome activation in the placentas of high BMI mothers, which inhibits placental insulin signaling and nutrient transport function. In vitro data suggests that placental inflammasomes are highly responsive to dietary fatty acids, with SFA and MUFA promoting inflammasome activation and n-3 PUFA inhibiting its activity. Future clinical studies are warranted to determine whether dietary interventions during pregnancy can impact upon infant and placental health by altering placental inflammatory pathways.
References
1.
2.
Catalano PM, Presley L, Minium J, Hauguel-de Mouzon S. Fetuses of obese mothers develop insulin resistance in utero. Diabetes Care. 2009;32:1076–80.PubMedCentralPubMedCrossRef
3.
4.
6.
Lumeng CN, Saltiel AR. Inflammatory links between obesity and metabolic disease. J Clin Invest. 2011;121:2111–7.PubMedCentralPubMedCrossRef
7.
Basu S, Haghiac M, Surace P, Challier JC, Guerre-Millo M, Singh K, Waters T, Minium J, Presley L, Catalano PM, Hauguel-de Mouzon S. Pregravid obesity associates with increased maternal endotoxemia and metabolic inflammation. Obesity (Silver Spring). 2011;19:476–82.CrossRef
8.
Aye IL, Lager S, Ramirez VI, Gaccioli F, Dudley DJ, Jansson T, Powell TL. Increasing maternal body mass index is associated with systemic inflammation in the mother and the activation of distinct placental inflammatory pathways. Biol Reprod. 2014;90:129.PubMedCentralPubMedCrossRef
9.
Basu S, Leahy P, Challier JC, Minium J, Catalano P, Hauguel-de Mouzon S. Molecular phenotype of monocytes at the maternal-fetal interface. Am J Obstet Gynecol. 2011;205(265):e261–8.
10.
Challier JC, Basu S, Bintein T, Minium J, Hotmire K, Catalano PM, Hauguel-de Mouzon S. Obesity in pregnancy stimulates macrophage accumulation and inflammation in the placenta. Placenta. 2008;29:274–81.PubMedCentralPubMedCrossRef
Related posts:
 Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
 Obesity and Inflammation in Pregnancy
Obesity and Inflammation in Pregnancy
 PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
 Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
 Maternal Diet, Developmental Origins, and the Intergenerational Transmission of Cardiometabolic Traits: A Window of Opportunity for the Prevention of Metabolic Syndrome?
Maternal Diet, Developmental Origins, and the Intergenerational Transmission of Cardiometabolic Traits: A Window of Opportunity for the Prevention of Metabolic Syndrome?
 Pathology of the Placenta: A Continuum Spectrum of Inflammation from Physiology to Disease
Pathology of the Placenta: A Continuum Spectrum of Inflammation from Physiology to Disease
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


