Fig. 6.1
Iron accumulation and ROS are important factors in the alcohol-mediated hepatocarcinogenesis
6.2 Health Statistics of ALD
Chronic alcohol consumption is one of the major risk factors worldwide, significantly affecting both mortality and years of life loss (YLL) [4]. Ca. 5 % of the Western world show risky alcohol consumption and some countries such as China reach a yearly regional increase of alcohol consumption of 400 % [5]. The liver is the major target organ of alcohol consumption, although alcohol may affect many other organ systems such as the heart, nervous system, pancreas, breast, and others. According to the recently published “burden of disease study 2010,” liver cirrhosis and liver cancer are ranked at position 12 and 16 in the actual death statistics [6]. In 2010 ca. one million people died from liver cirrhosis with one third directly attributable to alcohol. This is a considerable number when comparing with coronary heart disease with about ten million deaths and ranking as the leading cause of mortality in the global death statistics. In Central Europe, liver cirrhosis even ranks at the fourth position of YLL and HCC is now the most common fatal complication of patients with alcoholic liver cirrhosis. Moreover, it has the second fastest increase of all tumors worldwide after kidney tumors, and alcohol-associated HCC ranks on third position after HCC caused by viral hepatitis B and C infections. These epidemiological data explain the high interest in gaining a better understanding of molecular mechanisms of alcohol-driven hepatocarcinogenesis to identify novel target approaches for future therapies.
6.3 Prevalence of Iron Overload in ALD
A significant iron deposition in patients with chronic alcohol consumption has been appreciated for a long time [7] (see Fig. 6.2). Considerable confusion still exists today with genetic hemochromatosis, which is characterized by a typical and severe hepatocellular iron overload and shows other classical symptoms such as arthritis, skin pigmentation, and diabetes mellitus. The genetic nature of hereditary hemochromatosis was already clear when the first monographs on hemochromatosis in 1935 by Sheldon and the extensive review by Finch et al. in 1955 [8, 9] appeared. However, it was also noted [8] that patients with hemochromatosis showed a high prevalence of alcoholism of ca. 30 %. Although the introduction of percutaneous needle biopsy of the liver in the 1950s added a new tool for exact quantitation of hepatic iron deposition, it further led to considerable confusion since histological stainable iron deposits are common in cirrhotic livers [10, 11]. This is especially the case in alcoholic subjects and some investigators have even equated mild to moderate alcoholic cirrhosis with hemochromatosis. Notably, it is still not clear in what form the iron is actually deposited and in what form the iron is toxic (ferritin, hemosiderin, or labile iron pool [12, 13]). When analyzing the Finch data for differences between alcoholic and nonalcoholic patients, Powell noted that laboratory signs of hepatocellular damage showed a significant higher prevalence in the alcoholic group (serum transaminase elevation 35.5 % vs. 15.4 %). Interestingly, most large studies seem to indicate that mutations in the HFE gene [14] are not responsible for the iron accumulation [15–17] in ALD patients. Moreover, since hereditary hemochromatosis shows a very weak clinical penetration of about 10 %, it still remains open what exact role alcohol plays in patients with hemochromatosis and whether it is an important but underestimated disease modifier.
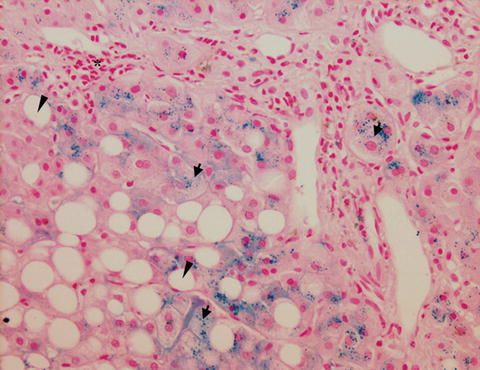
Fig. 6.2
Histological section of a liver biopsy from a patient with ALD. Staining of iron-loaded cells by Prussian blue (black arrows = intracellular inclusions of iron in hepatocytes) in the liver of ALD patient. Asterisk = inflammatory foci, arrow head = macrovesicular steatosis (fat droplet with displaced nucleus). In this patient, a predominant hepatocellular iron deposition is seen while Kupffer cells are not stained for iron
Important studies in the 1980s focused on pathologic abnormalities of available serum iron parameters. For instance, Chapman et al. determined the hepatic iron content in 60 alcoholics with liver disease and in 15 patients with untreated idiopathic hemochromatosis [18]. A significantly higher mean liver iron concentration was found in alcoholics as compared to controls (156.4 vs. 53 μg per 100 mg dry weight). However, liver iron was much higher in the hemochromatosis group (2,094.5 μg). In addition, no relationship between liver iron concentration and the amount of alcohol consumed was noted. Interestingly, Chapman also recognized that the serum ferritin concentrations reflected well the iron overload in patients with hemochromatosis and mild alcoholic liver disease while no association was found with serum ferritin in patients with severe alcoholic liver disease. He also concluded that serum iron and transferrin saturation were of little value in assessing iron status in patients with ALD. In 1994, Bell studied 312 patients with different liver diseases and, after a careful assessment of fasting serum iron, transferrin, and ferritin levels, found that serum ferritin is more frequently elevated in ALD than in other liver diseases [19]. In addition, measurement of ferritin should be postponed until the patients are abstaining because he noted that serum ferritin decreased from 1,483 to 388 μg/L after 1.5–6 weeks of abstaining from alcohol. The author claimed that most patients with increased serum ferritin had normal transferrin saturation. Again, ferritin levels higher than 1,000 μg/L were observed in 100 % of patients with hemochromatosis but only in 11 % in patients with ALD. Only 7.7 % of patients with other liver diseases had such high ferritin concentrations. It should be noted, however, that 15.2 % of ALD patients have increased transferrin saturation. In a recent study from Japan, excess iron accumulation was found in 22 hepatic tissues with alcoholic liver diseases, but not in any normal hepatic tissues [20].
We have recently characterized a large cohort at the Heidelberg Salem Medical Center using biopsy, but also various newer noninvasive tests including transient elastography. Selected data [21] from a cohort of 235 patients with ALD, of whom 86 had underwent a liver biopsy, with detailed iron characterization regarding intensity and location (macrophages vs. hepatocytes), are shown in Table 6.1. It can be summarized that ca. 50 % of patients with ALD have significant pathological iron deposition. Interestingly, iron deposits are found both at the same rate in macrophages and hepatocytes. In addition, there is a mixture of patient groups with only macrophages, only hepatocytes, and mixed iron deposition. It should be clearly noted that this iron deposition depends on fibrosis stage, as has already been seen many decades before. However, with new noninvasive technologies to better quantitate the stage of fibrosis (e.g., transient elastography), more quantitative associations can be shown (see Table 6.2). It should be also noted that the Heidelberger alcohol cohort shows a rather representative fibrosis distribution with about 60 % without any fibrosis, 10 % with fibrosis stages F1 and F2, 10 % fibrosis stage F3, and 15 % fibrosis stage F4. According to our data, the mean histological iron score (0–3) is 1 in patients with advanced fibrosis stage F3 and F4. About 2/3 of these patients with advanced fibrosis stage have elevated ferritin levels higher 400 μg/mL and an increased transferrin saturation higher in 45 %. This is especially important to consider, since ferritin levels and transferrin saturation are commonly used to screen patients with hereditary hemochromatosis. In the subanalysis (Mueller et al. 2013, unpublished data), we could show that the serum ferritin levels were highly associated with GGT, transaminases levels, transferrin saturation, liver stiffness, and histological iron deposition in hepatocytes and to a lesser extent to iron in macrophages. Longitudinal analysis of serum ferritin levels during abstaining from alcohol showed that concentrations decreased by 50 % within 2.1 days. Importantly, a careful comparison between patients with alcoholic liver disease and patients with nonalcoholic liver disease (NALD) in 30 cases matched with regard to gender, age, and fibrosis stage showed significant differences between the two cohorts with regard to iron-related proteins. So ALD patients showed lower transferrin levels and lower erythrocyte counts and hemoglobin levels but significantly higher ferritin level [22].
Table 6.1
Location and intensity of histological iron deposits in the ALD cohort from the Heidelberg Salem Medical Center
Parameter | Units | Mean | Fibrosis stage | Elevated (%) | ||
|---|---|---|---|---|---|---|
F0–2 | F3 | F4 | ||||
Ferritin (ng/mL) | m (30–400) | 567.2 | 678.2 | 1,062.0 | 509.2 | |
Transferrin (g/L) | (2–3.6) | 2.5 | 2.3 | 2.4 | 1.9 | |
Serum iron (ug/dL) | (59–158) | 157.0 | 115.1 | 90.6 | 83.2 | |
Cirrhosis (ultrasound) | 18.75 % | 6.52 % | 25.00 % | 64.71 % | ||
Liver stiffness (kPa) | kPa | 17.4 | 7.1 | 23.4 | 51.1 | |
Iron (macrophages) | 0–3 | 0.67 | 0.59 | 0.88 | 0.63 | 45.24 |
Iron (hepatocytes) | 0–3 | 0.61 | 0.57 | 1.00 | 0.38 | 42.86 |
Steatosis (Kleiner score) | 0–3 | 1.73 | 1.96 | 1.88 | 1.76 | 89.41 |
Lobular inflammation | 0–3 | 1.15 | 1.24 | 1.59 | 1.35 | 90.59 |
Ballooning | 0–2 | 0.72 | 0.67 | 0.94 | 1.00 | 63.53 |
Mallory–Denk bodies | 0–1 | 0.26 | 0.16 | 0.35 | 0.47 | 25.88 |
Cirrhosis (Kleiner) | 0–4 | 2.20 | 1.59 | 3.00 | 4.00 | 88.68 |
Table 6.2
Typical routine laboratory tests and ultrasound parameters in the actual Heidelberg cohort of ALD patients
Parameter | All (%) | No or low fibrosis (F0–2) (%) | advanced fibrosis (F3–4) (%) |
|---|---|---|---|
Laboratory | |||
GGT >60 IU/L | 75.0 | 61.9 | 95.2 |
GOT/GPT >1 | 81.3 | 77.1 | 92.9 |
GOT >50 IU/L | 60.1 | 51.4 | 88.1 |
GPT >50 IU/L | 46.5 | 41.6 | 66.7 |
Ferritin >400 ng/mL | 37.3 | 28.6 | 66.7 |
Transferrin saturation >45 % (%) | 36.1 | 27.8 | 55.3 |
Bilirubin >1.3 mg/dL | 15.8 | 5.7 | 26.2 |
Ultrasound | |||
Sonographic signs of cirrhosis (%) | 19.6 | 1.0 | 40.0 |
Splenomegaly (>11 cm) (%) | 11.4 | 0.0 | 15.0 |
Ascites (%) | 25.7 | 17.6 | 32.3 |
Taken together, patients with ALD show a significantly increased histological iron deposition in about half of the population that increases with disease progression toward cirrhosis (see Fig. 6.3). This iron deposition is reflected by serum ferritin levels, but unfortunately overlayered by additional inflammatory conditions. In fact, according to our analysis, serum ferritin levels reflect hepatocyte iron deposition more closely than macrophage iron deposition in ALD. Moreover and often overlooked, a significant proportion of patients show elevated levels of transferrin saturation and ferritin levels that can be easily mistaken for hemochromatosis.
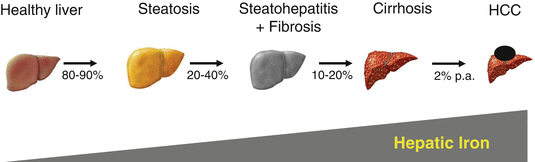
Fig. 6.3
Natural course of ALD ultimately leading to liver cancer
6.4 Iron Toxicity and Carcinogenesis
Iron toxicity has been well known for many decades and usually attributed to Fenton chemistry, first described in 1896 by Fenton and referring to the reaction of reduced ferrous iron with H2O2 to ferric iron and hydroxyl radicals [23]. These radicals are highly and universally reactive to all cellular compounds including DNA, proteins, lipids, and carbohydrates. They cause oxidative damage to DNA, including base modifications and DNA strand breaks, and may change the structure of proteins and lipids, eventually leading to mutagenesis.
Although, the direct chemical nature of the iron toxicity in Fenton’s chemistry has been recently questioned by Saran et al. [24], it is generally assumed that at least Fenton-like reactions contribute to the toxicity of iron. For this reason, we will not further differentiate both reactions in this review. In vitro studies have shown that liver viability decreases in the presence of high amounts of iron and that the depletion of iron by iron chelators can protect cells from peroxide toxicity. Hepatic iron overload and the risk for liver cancer have also been extensively studied using in vivo experiments in the past. In patients with hereditary hemochromatosis due to HFE mutations, the hepatic iron overload ultimately determines the overall survival [25]. In a large transplant registry study, Ko et al. showed that iron alone increased the risk of HCC by a factor of 2.2 [26]. Turlin et al. showed in 1995 that the iron-associated risk of HCC was not dependent on whether the patient had cirrhosis or not [27]. In an interesting study from the NIH, Zacharski et al. could demonstrate that iron depletion by phlebotomy may have a protective effect on general carcinogenesis [28]. He studied prospectively 641 controlled subjects without iron depletion and 636 subjects in an iron reduction group. Iron reduction was achieved by monthly phlebotomy. Patients were followed up for an average for 4.5 years. Interestingly, the risk of new visceral malignancies was lower in the iron reduction group than controlled group (38 vs. 60), and among patients with new cancers, those in the iron reduction group had lower cancer-specific and all-cause mortality. Mean ferritin levels across all 6-monthly visits were lower among all patients who did not develop cancer. Notably, in a 1988 animal study, Hann et al. showed reduced tumor growth in iron-deficient mice [29], and Tsukamoto et al. in 1995 showed in rodents that a mixture of high fat diet and alcohol together with supplemented iron led to pronounced cirrhosis [30]. In a meta-analysis by D’Amico et al., it was concluded that liver iron is one among several independent prognostic factors of survival in liver cirrhosis [31]. Ganne-Carrie showed in 229 patients with ALD or hepatitis C virus-related cirrhosis that iron is the best parameter for predicting mortality in ALD patients (p = 0.007) followed over 57 months [32]. This study found no prognostic significance of hepatic iron for HCV.
Taken together, there seems to be enough evidence from human and animal studies to suggest that hepatic iron overload leads to hepatocellular damage. This in turn leads to increased fibrosis progression and risk for hepatocarcinogenesis, and ultimately iron seems to be an important prognostic factor for overall survival in patients with ALD. The most evident and generally accepted mechanisms of iron carcinogenesis include Fenton-like reactions that lead to the highly aggressive hydroxyl radicals.
6.5 Mechanism of Iron Overload
Enormous progress has been made in the last two decades to better understand the molecular mechanisms of iron regulation and homeostasis both at the cellular and systemic level [33–35]. These milestones include the discovery of iron regulatory proteins (IRP) and their posttranscriptional regulation of cellular iron homeostasis in the late 1980s and early 1990s as well as identification of the systemic master switch peptide hepcidin about 10 years ago [36]. Despite this progress, numerous aspects of iron regulation still remain unexplained in humans. Apart from the diagnosis of rare and common variants of hereditary hemochromatosis, the progress has led to significant improvements in the diagnosis and therapy of iron overload. The discovery of the HFE gene that causes the majority of hereditary hemochromatosis forms has clearly helped to resolve the role of hemochromatosis in alcoholics [14]. Interestingly, most large studies seem to indicate that mutations in the HFE gene [14] are not responsible for the iron accumulation in patients with ALD [15–17]. These studies thus seem to rule out HFE as important genetic disease modifier. In contrast, as briefly discussed above, alcohol could be an important disease modifier in patients with hemochromatosis, but typically with weak clinical penetration. In addition, at the systemic level, ALD appears rather complex since alcohol affects not only the liver but also many other organ systems including the bone marrow and the immune system so that iron regulation obviously is altered at many different levels and locations throughout the body. In the following, a brief description of the cellular and systemic iron homeostasis is given and the status quo of molecular findings in patients with ALD is discussed.
6.6 Systemic Regulation
The majority of body iron, roughly 2 g in humans, is distributed in the oxygen carrier hemoglobin of red blood cells and developing erythroid cells (Fig. 6.4). Excess iron is usually stored in the liver, which normally contains about 1 g of iron, predominantly in the form of ferritin [34]. Ferritin is a protein composed of 24 subunits with a ferrihydrite core where 4500 iron atoms can be stored in cage-like structures. It consists of ferritin H chain (heavy or heart) and ferritin L chain (liver or light) subunits and can be regulated by IRPs [37]. Other significant amounts of iron are found in macrophages (0.6 g) and in the myoglobin of muscles (0.3 g). It is generally believed that mammals lose iron from regular sloughing of the mucosa and skin cells or during bleeding and do not possess any mechanisms for iron excretion from the body [33]. Therefore, the balance is maintained by the tight control of dietary iron absorption at the brush border of enterocytes in the proximal duodenum (Fig. 6.4). Dietary iron uptake involves the reduction of ferric iron to ferrous iron in the intestinal lumen by ferric reductases, such as duodenal cytochrome B (Dcytb), and subsequent transport of iron via the apical membrane of enterocytes by divalent metal transporters (such as DMT1 encoded by SCL11A2 (solute carrier family 11, member 2) gene). Iron can also be transported across the enteral membrane in the form of heme by unknown mechanisms and the iron is then released inside enterocytes via heme oxygenase 1 (HO-1). Cytosolic iron is exported by the basolateral iron transporter ferroportin (FP-1 or SLC40A1) from the enterocytes to the blood compartment and then bound to transferrin, the major abundant iron-carrying protein within serum. Before binding, ferrous iron is oxidized by ferroxidases (e.g., hephaestin/ceruloplasmin) to the ferric form. Iron is then distributed within the body and used in various pathways, but mainly utilized in the bone marrow for the synthesis of new heme.
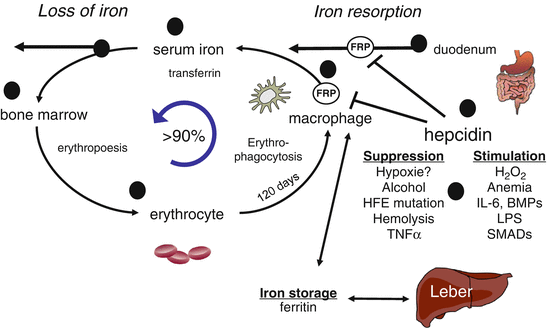
Fig. 6.4
Systemic iron homeostasis and utilization in the body. Black circles indicate potential target sites of iron regulation affected by chronic or acute alcohol consumption. Dietary iron is absorbed in the duodenum and bound to transferrin. Iron is then mainly delivered to the bone marrow for erythropoiesis while senescent erythrocytes are phagocytosed by macrophages. This efficient mechanism recycles >90 % of all iron for new heme synthesis. Excess iron is stored in hepatic ferritin or temporarily in macrophage ferritin. Regulation of iron metabolism by hepcidin and upstream factors are also shown
About 1–2 mg per day of iron is absorbed to keep a stable iron balance. Of note, iron undergoes an efficient recycling. Senescent erythrocytes that have a life span of 120 days are sequestered by macrophages and the iron is reused for new heme synthesis. This recycling machinery accounts for about 90 % of newly synthesized hemoglobin. It is important to note that the iron export pump, ferroportin, is not only found on enterocytes but also on macrophages and hepatocytes (Fig. 6.5). The ferroportin-mediated efflux of ferrous iron from enterocytes and macrophages into the serum is critical for systemic iron homeostasis and mainly controlled via the liver-secreted 25 AS peptide hormone, hepcidin. Mechanistically, hepcidin binds to ferroportin and promotes its phosphorylation, internalization, and lysosomal degradation [38, 39]. Hepcidin is primarily expressed in hepatocytes as a precursor pro-peptide, although other locations of secretion have been described, such as macrophages [40] (Fig. 6.5) and to a lesser extent cardiomyocytes. Hepcidin efficiently blocks ferroportin, which leads to accumulation of iron within macrophages and blocks the iron absorption via enterocytes (Fig. 6.4).
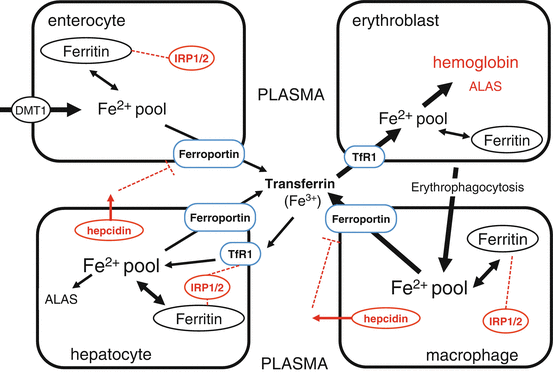
Fig. 6.5
Interaction of systemic and cellular iron homeostasis at four major sites: enterocyte, hepatocyte, erythroblast, and macrophage
An important evolutionary conserved mechanism to induce hepcidin is an infection or inflammatory state. Via cytokines, namely, IL-6, but also microbial molecules (such as lipopolysaccharide), hepcidin is strongly induced leading to a rapid decrease of serum iron, which is thought to function as an antibacterial defense mechanism. More recently, another important inflammatory cofactor, H2O2, has been identified as potent inducer of hepcidin. In contrast, hepcidin levels seem to be suppressed in patients with genetic hemochromatosis, leading to increased uptake of iron via the duodenum and increased release of iron through macrophages. Cytokine-mediated induction of hepcidin is thought to be the reason for anemia of chronic disease [41], while the disruption of hepcidin is generally associated with the systemic iron overload (e.g., genetic hemochromatosis). Despite the progress and the discovery of various upstream regulators of hepcidin (such as C/EBPα; BMP6; SMAD 1, 5, 8, and 4; TMPRSS6; IL-6; CREBH; CHOP; and TLR4), an overall and conclusive understanding of the regulatory network with respect to the control of iron is not yet completely understood. Hitherto unexplained features of hepcidin regulation include:
1.
Expression of hepcidin in cells other than hepatocytes
2.
The nature of co-expressing ferroportin in hepatocytes and macrophages
3.
The experimental and clinical finding that hepcidin responds differentially to iron overload in vitro and in vivo
4.
The controversial findings of hypoxia-mediated regulation of hepcidin
5.
The controversial and conflicting findings of the response of hepcidin toward ROS
At least to the latter point we could recently identify a partial explanation [42]. Thus, the central ROS, H2O2 induces hepcidin in hepatocytes independent of IL-6 when exposed in continuous manner. Bolus treatments, however, which reflect an artificially high H2O2 exposure blocked hepcidin expression [43]. They are even toxic at H2O2 concentrations higher than 50 μmol and hepcidin suppression at such conditions was due to unspecific inhibition of the mRNA transcription machinery [43].
6.7 Cellular Iron Regulation
Developing erythrocytes, as well as most other cell types, require iron from plasma transferrin. Iron is loaded onto transferrin with a capacity of two atoms of ferric iron per molecule. Transferrin binds with a very high affinity to cell surface transferrin receptor 1 (TfR1) ubiquitously expressed and with much lower affinity to transferrin receptor 2 (TfR2) sharing 45 % homology to TfR1 [44, 45] (see Fig. 6.6). Homology aside, TfR2 exhibits different properties from TfR1. TfR2 is only expressed in liver and to a lesser extent in erythroid, spleen, lung, and muscle cells. Also, its mRNA levels are not directly regulated by cellular iron content. Its expression is higher in iron-replete conditions, suggesting a role as transferrin-iron sensor candidate rather than acting primary in iron uptake [46]. The TF-TfR1 complex is endocytosed via clathrin-coated pits. The release of ferrous iron into the intracellular compartment requires acidification of the endosome using a proton pump and the reduction of ferric iron to the ferrous form via Steap3. DMT1 transports the ferrous iron across the endosomal membrane into the cytosol where it can be stored in ferritin complexes in non-erythroid cells or incorporated into hemoglobin in erythroid cells [47] (see Fig. 6.6). Iron is then either reused for various synthesis pathways such as intracellular heme synthesis and iron cluster protein synthesis or transported into the major iron storage protein ferritin.
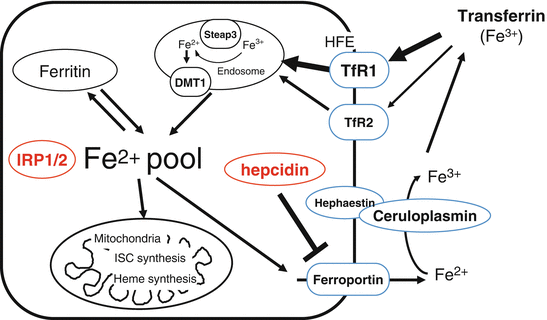
Fig. 6.6
Cellular iron metabolism. Iron-loaded holo transferrin binds to its cell surface receptor (high affinity TfR1, low affinity TfR2) and the Tf-TfR complex is endocytosed. In the endosome, iron is released and reduced by Steap3 and transported across the endosomal membrane by DMT1. Internalized iron is directed to mitochondria for heme or ISC synthesis and excess iron is stored in ferritin. Cytosolic iron is exported by the iron transporter ferroportin to the blood compartment and then again bound to transferrin. Regulatory mechanisms by hepcidin or IRPs are shown in red
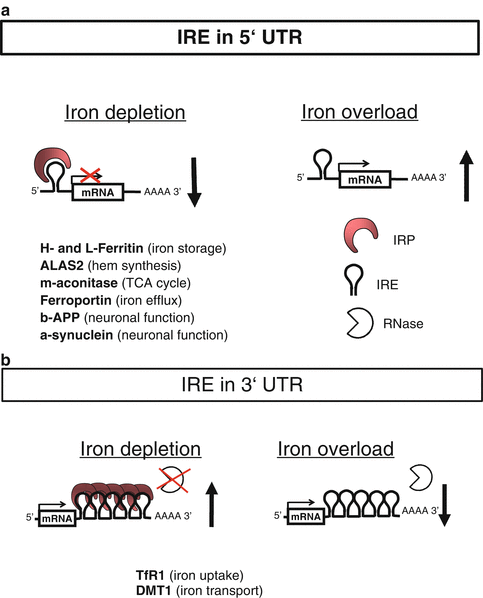
The expression of DMT1 and ferritin is coordinately posttranscriptionally regulated by binding of trans-acting iron-responsive proteins (IRP1 and IRP2) to iron-responsive elements (IRE) in the untranslated regions (UTRs) of their respective mRNAs [48–50]. In iron-starved cells IRPs bind with high affinity to cognate IREs (see Fig. 6.7a, b). IREs are evolutionary conserved hairpin structures of about 30 nucleotides with characteristic sequences. The effect of IRP binding to IREs is dependent on their position. TfR1 mRNA contains five IREs within its long 3′ UTR that stabilizes and protects the transcript from degradation leading to protein upregulation, but other mRNAs, for example, mRNAs encoding H- and L-ferritins, contain a single IRE in their 5′ UTRs where binding results in decreased protein translation by steric blockade. As a result increased TfR1 levels stimulated acquisition of iron from plasma transferrin to counteract iron deficiency. The inhibition of ferritin synthesis leads to decreased abundance of this protein as iron storage becomes obsolete under these conditions. Conversely, in cells with high iron content, both IRP1 and IRP2 become unavailable for IRE binding lowering TfR1mRNA degradation and ferritin mRNA translation. Thus, when iron supply exceeds cellular means, the IRE-IRP switch minimizes further iron uptake via TfR1 and favors the storage of excess iron in newly synthesized ferritin. Other IREs have been discovered in the genes of ALAS2, mitochondrial aconitase, ferroportin, HIF2α, β-APP, and α-synuclein, which in turn control iron storage and erythroid iron utilization, energy homeostasis, hypoxia responses, and neurological pathways, respectively.
 A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
 Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
 Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
 Alcohol and HCV: Implications for Liver Cancer
Alcohol and HCV: Implications for Liver Cancer
 Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury




Related posts:
A Perspective on Chemoprevention by Resveratrol in Head and Neck Squamous Cell Carcinoma
Transgenic Mouse Models for Alcohol Metabolism, Toxicity, and Cancer
Genetic Polymorphisms of Alcohol Dehydrogense-1B and Aldehyde Dehydrogenase-2, Alcohol Flushing, Mean Corpuscular Volume, and Aerodigestive Tract Neoplasia in Japanese Drinkers
Alcohol Consumption, Wnt/β-Catenin Signaling, and Hepatocarcinogenesis
Alcohol and HCV: Implications for Liver Cancer
Synergistic Toxic Interactions Between CYP2E1, LPS/TNFα, and JNK/p38 MAP Kinase and Their Implications in Alcohol-Induced Liver Injury
Stay updated, free articles. Join our Telegram channel
Full access? Get Clinical Tree
