Fig. 5.1
Pathways indicating the integrative function of estrogen in regulating glucose uptake. Estrogen mediates glucose homeostasis via its two receptors ERα and ERβ. ERα, the predominant isoform in muscle, adipose, and liver tissues has genomic and non-genomic mechanisms regulating insulin action and glucose metabolism. ER estrogen receptor, p phosphorylated, FAO fatty acid oxidation, OXPHOS oxidative phosphorylation, FFA free fatty acids, AMPK AMP-dependent kinase, NFkB nuclear factor kappa B, HSP heat shock proteins
The ERs also play a role in glucose metabolism, with ERα and ERβ knockout (KO) mice providing much of the initial understanding of ER regulation of glucose control. Increased adiposity occurs in humans and mice as a result of decreased ERα activation [23, 54], and mice with global knockout of ERα exhibit impaired glucose tolerance and skeletal muscle insulin resistance [54–56]. Based on this evidence, the beneficial effects of estrogens on glucose metabolism are thought to be mediated by ERα.
Mechanisms of ER-Mediated Glucose Metabolism
While activation of the estrogen receptors has the potential to positively modulate glucose metabolism, the exact mechanism of action is unknown. Some studies suggest a mechanism by which estrogen receptors modulate GLUT4, the critical glucose transporter in skeletal muscle. NF-κB is a transcription factor that is activated by stimuli such as cellular stress, cytokines, and inflammation. The promoter region of GLUT4 contains an NF-κB binding site [57], and NF-κB represses GLUT4 transcription [58]. In a basal state, NF-κB is bound by the inhibitor of kappa B α (IκBα) in the cytosol and remains inactive. Upon activation of the stress kinase proteins, IκBα is phosphorylated, which signals its degradation by the proteosome. The free NF-κB is then activated and translocates to the nucleus where it functions as a transcription factor. Tumor necrosis factor-α (TNF-α) is a cytokine that activates the NF-κB pathway and is highly expressed in obese humans [59, 60]. In addition, obese humans with T2DM have increased skeletal muscle NF-κB activation [61]. Previous studies have also shown that rats on an obesity-promoting high-fat diet have decreased skeletal muscle GLUT4 protein [62–64]. Overall, these studies suggest that obesity increases the amount of TNF-α and leads to NF-κB activation, which is followed by a decrease in GLUT4 protein levels.
New evidence suggests NF-κB, and Glut4 consequentially, is regulated by ERα. Rather than binding directly to DNA, ERα can also modulate gene expression by binding transcription factors [65–67]. Previous studies have shown that activated ERα can directly bind to NF-κB and decrease NF-κB–DNA binding [65, 68–70]. Activated ERα could essentially serve a protective function in regards to GLUT4 expression. The opposite would also be true, that low levels of activated ERα may result in decreased GLUT4 levels. In fact, females with polycystic ovarian syndrome who have high androgen and low estrogen levels (and, therefore, low ERα activation) have 35 % less GLUT4 protein compared to control females [71]. In addition, ERα KO mice show a decrease in GLUT4 mRNA levels [72]. Therefore, increased NF-κB activation via a high-fat diet and obesity combined with low ERα activation could decrease GLUT4 transcription, leading to a subsequent decrease in glucose uptake and insulin resistance. This physiological condition (obesity and low E2 levels) is present in most postmenopausal women, putting these women at a particular risk for insulin resistance.
Another postulated mechanism for estrogen-related protection of glucose homeostasis is the mitigation of inflammation and oxidative stress that are implicated in causing insulin resistance. ERα deficient mice have high levels of Plasminogen activator inhibitor-1 (PAI-1) and TNF-α, markers of inflammation, and reduced adiponectin, a suppressor of inflammation and inducer of insulin sensitivity [73]. Muscles of ERα knockout mice also exhibit greater proinflammatory lipid breakdown products diacylglycerol and ceramides, along with increased activation of stress kinases like JNK, all of which have been implicated in inducing insulin resistance in skeletal muscle. The mechanism by which ERα mitigates the proinflammatory state remains to be completely understood. One mechanism could include the ability of ERs to regulate fatty acid oxidation and mitochondrial function. Previous studies indicate that ERα can modulate mitochondrial function, including ATP production, mitochondrial membrane potential, and calcium concentrations [74, 75]. ERα directly interacts with mitochondrial fatty acid oxidation enzyme HADHB. 17-β Estradiol increases the activity of HADHB in wild-type cells but not in cells lacking ERα [76]. ERα is thought to have direct positive transcriptional effects on increasing mitochondrial biogenesis [77], and ERα binding sites are enriched in the promoters of genes involved in energy metabolism [78]. Recent studies also show the presence of ERs within the mitochondrial membrane [79–81]. Mitochondrial ERs can bind directly to estrogen receptor elements, suggesting that ERs may be directly involved in estrogen-induction of mtDNA transcription [82]. Therefore, with a coordinated action of nuclear ERs, mitochondrial ERs, and their coactivators, estrogen signaling may regulate mitochondrial function in tissues with high energy demand, like skeletal muscles. Increased mitochondrial oxidative phosphorylation and mitochondrial fatty acid oxidation improves complete fatty acid metabolism, preventing the buildup of proinflammatory intermediates like DAG and ceramides [83]. Reduction in DAG and ceramides improves glucose metabolism by reducing activation of stress kinases that are known to inhibit the insulin-signaling pathways.
Several additional mechanisms have been postulated by which estrogens can regulate glucose metabolism. Estrogens may improve insulin action by mitigating oxidative stress [84] and reducing mitochondrial ROS production [85]. Another mechanism by which ER can regulate glucose metabolism is the activation of PPARγ, a master regulator of insulin/glucose metabolism. ERβ inhibits ligand-mediated PPARγ-transcriptional activity to reduce adipogenesis, while high-fat diet-fed ERβ knockout mice are more insulin sensitive despite having more adiposity [86]. Packaging of fatty acids in adipose depots such that they become incapable of inhibiting insulin signaling in muscle tissue is an important mechanism of PPARγ ligands in maintaining glucose homeostasis. Estrogen may also affect glucose homeostasis by regulating muscle mass, which are the primary glucose disposal centers in the body. In L6 cells, estrogen treatment increases GLUT4 translocation to the membrane, along with expression of myogenesis markers, including myogenin and MHC [21]. Estrogen-dependent myogenesis was prevented by translation inhibitor cycloheximide and transcription inhibitor actinomycin, suggesting that nuclear mechanism of ER action was involved in muscle differentiation. The varied effects of estrogen on glucose metabolism are only now being uncovered and are not yet fully understood.
Estrogen Regulation of Glucose in Insulin-Sensitive Tissues
Glucose regulation in the body mostly occurs in the skeletal muscle, adipose tissue, and liver (Fig. 5.2). ERs are also present in the brain and have profound effects on food intake [87]. ERs are thought to play a critical role in the regulation of glucose metabolism by estrogen in each of these metabolic tissues.
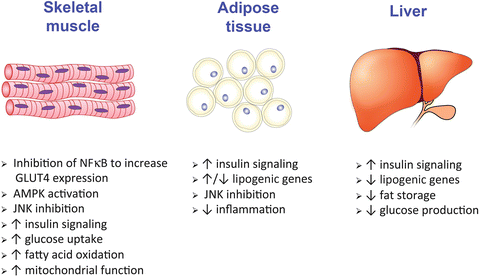
Fig. 5.2
Differential effects of ERα activation on glucose and fatty acid metabolism in skeletal muscle, adipose tissue, and liver. AMPK AMP-dependent kinase, NFkB nuclear factor kappa B, JNK c-Jun N-terminal kinase. Image credit to Stanton Fernald, KIDDRC Illustration &Imaging Center, University of Kansas Medical Center
Estrogen Regulation of Glucose Metabolism in Skeletal Muscle
Approximately 75 % of glucose regulation in the body occurs in skeletal muscle [88] and estrogen receptors are highly expressed in skeletal muscle. In mice that do not express ERα, a decrease in whole body glucose tolerance and a decrease in glucose uptake into the skeletal muscle is observed [54, 56, 89]. Even on a normal chow diet, a lack of ERα resulted in impaired glucose tolerance and reduced insulin sensitivity in skeletal muscle and liver [89]. Whether the whole body changes can be attributed to impaired insulin action in skeletal muscle or the liver is not clear [56].
Previous findings from our laboratory demonstrate an increase in insulin-stimulated glucose uptake with PPT, suggesting an important role for ERα in mediating skeletal muscle insulin sensitivity [90]. While ob/ob mice treated with PPT for 7 days demonstrated no increase in glucose uptake in soleus or EDL muscle [91], this could be attributed to the significantly lower dose of PPT used in the latter study. It can be somewhat difficult to parcel out the effects of ERs in skeletal muscle glucose regulation, however, given the fact that estrogen stimulates both ERα and ERβ. It is likely that estrogens and estrogen receptor modulators produce distinct phenotypes depending on whether a tissue expresses predominately ERα or ERβ [92]. It has been suggested that ERα is more highly expressed in insulin-sensitive tissues [51, 89, 93, 94]. However, other accounts indicate that ERβ expression predominates in skeletal muscle [95]. ERα and ERβ regulation of function varies with the target tissue and activation of ERβ could oppose the action of ERα in the regulation of glucose metabolism [47, 72, 96]. Distinct tissue-specific effects of estrogen and estrogen receptor modulators occur with the recruitment of different coregulatory proteins to ERs [92, 97]. Differences in coregulatory protein recruitment can result in a modulator expressing agonist or antagonist properties. The coregulatory proteins involved in ERα activation by estrogen and PPT in skeletal muscle have not yet been identified, but could have an important role in determining the regulation of glucose metabolism by estrogen in vivo.
While studies demonstrate that estrogen results in increased insulin signaling, this does not appear to have a functional effect on glucose uptake in vivo. Phosphorylation of Akt [98] and AMPK [99] occurs with estrogen treatment in C2C12 muscle cells, and acute incubations (5 and 10 min) with estrogen increase phosphorylation of Akt, AMPK, and TBC1D1/4 in soleus muscle [100]. However, insulin-stimulated glucose transport is not increased with acute in vivo estrogen incubation. In addition, 3 days of estrogen treatment in vivo demonstrated no effect on insulin-stimulated glucose uptake in soleus or EDL muscles [90]. This is in contrast to an increase in glucose uptake with 3 days of with PPT treatment. The activation pattern of ERs with estrogen in vivo remains to be determined in skeletal muscle.
When ERα is stimulated via PPT, direct activation of the insulin-signaling pathway occurs as shown by increased phosphorylation of Akt [90]. In C2C12 myotubes, stimulation of the insulin-signaling pathway with resveratrol was also shown to be dependent on ERα activation [101]. ERα also has an effect on insulin-independent pathways of glucose regulation with PPT increasing phosphorylation of AMP-dependent kinase (pAMPK) in soleus and EDL muscles [90]. ERα-KO mice demonstrate decreased pAMPK in skeletal muscle [89] and a recent study demonstrates that estrogen-induced AMPK activation is mediated by ERα [100]. These findings suggest that ERα acts as a positive modulator of AMPK activation. AMPK can phosphorylate both AS160 [102, 103] and TBC1D1 [104, 105], although with phospho-specific sites distinct from those activated by Akt, to stimulate an increase in glucose uptake. The ability of ERα to stimulate both insulin-dependent and non-insulin-dependent pathways has important implications for the regulation of glucose uptake in vivo.
ERα may also regulate glucose metabolism through direct modulation of GLUT4. GLUT4 is decreased in the gastrocnemius muscle of male ERα knockout (ERα-KO) mice [72]. However, a more recent report shows no change in GLUT4 levels in the quadriceps or soleus muscles in female ERα-KO mice [89]. It is possible that the ability of ERα to regulate GLUT4 may be fiber type specific as Gorres et al. demonstrated that activation of ERα results in increased GLUT4 in the EDL (fast-twitch) but not in the soleus (slow-twitch) muscle [90]. While numerous transcriptional pathways regulate GLUT4 [106], acute ERα activation may be an additional and lesser known mechanism for modulating GLUT4.
Estrogen Regulation of Glucose Metabolism in Adipose Tissue
Studies show that estrogen treatment in adipocytes increases insulin-stimulated glucose uptake and activation of the insulin-signaling pathway more than insulin alone [107, 108]. Furthermore, Muraki et al. found that the beneficial effects of estrogen were abolished when adipocytes were co-treated with methylpiperidinopyrazole (MPP), a specific ERα inhibitor. The beneficial effects were restored with PPT treatment to activate ERα [107]. These studies suggest that activation of ERα can potentiate the insulin-signaling pathway and glucose uptake in cultured adipocytes.
In adipose tissue, ERα KO mice have increased body weight and white adipose tissue weight compared to WT mice. ERα KO mice also have increased adipocyte size and number, although food intake does not differ [54]. Similarly, aromatase KO mice, in which androgens cannot be converted to E2, have increased body weight [109] and adipose tissue weight [110] compared to WT mice. In contrast, ERβ KO mice do not have increased adipose tissue weight or percent body fat compared to WT mice [111]. Therefore, estrogen/ERα signaling appears to be an important regulator of body weight and adipocyte regulation. Ovariectomy of ERα KO mice improves insulin resistance after reducing adipocyte size, indicating a role of ERβ in negative regulation of insulin action. The relative ratio of ERα/ERβ expression seems to be directly associated with obesity as well as serum level of leptin in omental adipose tissue of women [112].
Estrogen has the potential to regulate fat storage and triacylglyceride accumulation by altering transcription of lipogenic proteins such as SREBP-1 and its downstream targets, ACC, and FAS [56, 113–116]. The effects of estrogen on lipogenic pathways have primarily been assessed in response to estrogen treatment or replacement. For example, Phrakonkham et al. [117] demonstrated that estrogen treatment increased FAS expression in cultured adipocytes. However, other studies have demonstrated opposite effects, with estrogen treatment in mice shown to decrease ACC and FAS mRNA in adipose tissue [114, 118]. As has been previously shown, physiological estrogen levels may positively modulate glucose metabolism, while high or low estrogen levels have a different effect [107, 108]. More studies are needed to assess the role of estrogen and ER expression in modulating lipogenic pathways in cycling, OVX, and estrogen- treated animals.
Increased lipid intermediates and oxidative stress in insulin-responsive tissues can result in activation of stress kinases [119–123]. We [119, 120] and others [89, 124] have previously shown that increased stress kinase activation and decreased heat shock protein (HSP) expression contribute to decreased insulin signaling and glucose uptake in skeletal muscle. Further evidence suggests that ERα may be involved in stress kinase activation and HSP expression. Ribas et al. demonstrate increased activation of JNK in skeletal muscle and adipose tissue of ERα knockout mice [89]. When challenged with a high-fat diet, ERα knockout mice display greater JNK activation and decreased HSP72 expression in adipose tissue compared to high-fat fed wild-type mice [89]. These data suggest that ERα may contribute to glucose regulation by positively modulating stress kinase activation and HSP expression.
Estrogen Regulation of Glucose Metabolism in the Liver
With respect to the liver, ERα KO mice show hepatic insulin resistance during the euglycemic-hyperinsulinemic clamp test [56]. While hepatic glucose production decreases in wild-type (WT) mice, insulin is not able to decrease hepatic glucose production in ERα KO mice. Gene analyses of liver tissue from ERα KO mice exhibit downregulation of genes regulating lipid transport and upregulation of genes for hepatic lipid synthesis. Estrogen treatment reduces hyperglycemia, oxidative stress, and ameliorates liver dysfunction in diabetic rats via increased expression and signaling of insulin receptors [125].
ERs have a profound role in lipid metabolism in the liver. In liver, ERα has direct binding sites on promoters of genes involved in lipid and glucose metabolism, including PPARα, PDK4, PCK1 [78]. The anti-diabetic effects of estrogen treatment to diabetic ob/ob mice are associated with reduced lipogenic genes in the liver [126]. On the other hand ERβ KO mice have normal hepatic glucose output and insulin secretion [56], suggesting that ERα likely plays the predominant role in regulation of hepatic glucose homeostasis, possibly due to the much greater expression of ERα in the liver compared to ERβ. But another report showed that ERβ KO fed a high-fat diet have decreased liver fat and increased insulin sensitivity, possibly due to the reduction in triglyceride accumulation in the liver [86].
Estrogen treatment to ovariectomized mice protects against fatty liver disease when fed a high fat diet [127]. The insulin-resistant liver faces a paradox for insulin action because insulin resistance impairs the ability of insulin to suppress gluconeogenesis but increases insulin’s ability to promote lipogenesis. Estrogen treatment selectively inhibits the lipogenic aspect of insulin signaling and yet promotes the glucose suppressing-action of insulin [127], maintaining overall glucose homeostasis in the body in the face of a HFD.
Recent studies indicate that amount of fat accumulation in the liver is a better predictor of complications of obesity than visceral fat including insulin resistance and diabetes. Estrogen and ERα agonists have been associated with reduced fatty liver disease in mouse models [128] and women [129]. Women with fatty liver disease have hyperinsulinemia and insulin resistance, but also have less estrogen levels than women without fatty liver disease, concurrent with the observation that fatty liver disease is more prevalent in postmenopausal and women with polycystic ovarian syndrome [129].
In summary, estrogen mediates its metabolic effects on tissue via its two receptors, ERα and ERβ; and many of these effects are dependent on the expression levels and activation of the two receptors in various tissues. In insulin-sensitive tissues like skeletal muscle, adipose tissue, and liver, ERα is the predominant mediator of estrogen action. Activation of ERα increases energy metabolism pathways, enhances efficient fatty acid oxidation and improves insulin sensitivity. Estrogen’s metabolic effects are achieved via both genomic mechanisms, for example increasing expression of genes for mitochondrial biogenesis, and via non-genomic mechanisms, for example prevention of NFκB-mediated GLUT4 inhibition. In skeletal muscle, estrogens increase insulin signaling and glucose uptake, while in the liver, estrogens reduce fat storage and suppress glucose production. In contrast, estrogens may promote fat storage in adipose tissue in order to secure excess fatty acids which could be harmful in the circulation.
Conclusion
Estrogen’s regulation of glucose metabolism is a delicately balanced process in the body. While several reports show strong evidence of protective effects of estrogens, other studies clearly indicate that estrogen levels out of the physiological range, either lower or higher, are related to increased insulin resistance [130, 131]. Given the potentially contrasting roles of ERα and ERβ in regulating glucose homeostasis, ER isoform-specific agonists may have more beneficial effects in preventing insulin resistance in postmenopausal women than estrogen treatment alone. Future research and long-term clinical trials are required to verify the therapeutic benefits of ER modulators on glucose metabolism. The rapid increase in obesity and diabetes worldwide warrants extensive investigation of ERs to improve glucose homeostasis.
Acknowledgments
This work was supported by grants P20 RR016475 from the National Center for Research Resources (NCRR) and AG031575 from the National Institutes of Health, and AHA grant 12POST9020018 to A. A. Gupte.
References
1.
Expert Panel on Detection, Evaluation, and Treatment of High Blood Cholesterol in Adults (2001) Executive Summary of The Third Report of The National Cholesterol Education Program (NCEP) Expert Panel on Detection, Evaluation, And Treatment of High Blood Cholesterol In Adults (Adult Treatment Panel III). JAMA 285(19):2486–2497
2.
Park YW, Zhu S, Palaniappan L, Heshka S, Carnethon MR, Heymsfield SB (2003) The metabolic syndrome: prevalence and associated risk factor findings in the US population from the Third National Health and Nutrition Examination Survey, 1988–1994. Arch Intern Med 163(4):427–436PubMed
3.
Lindheim SR, Buchanan TA, Duffy DM, Vijod MA, Kojima T, Stanczyk FZ, Lobo RA (1994) Comparison of estimates of insulin sensitivity in pre- and postmenopausal women using the insulin tolerance test and the frequently sampled intravenous glucose tolerance test. J Soc Gynecol Investig 1(2):150–154PubMed
4.
Lynch NA, Ryan AS, Berman DM, Sorkin JD, Nicklas BJ (2002) Comparison of VO2max and disease risk factors between perimenopausal and postmenopausal women. Menopause 9(6):456–462PubMed
5.
Nuutila P, Knuuti MJ, Maki M, Laine H, Ruotsalainen U, Teras M, Haaparanta M, Solin O, Yki-Jarvinen H (1995) Gender and insulin sensitivity in the heart and in skeletal muscles. Studies using positron emission tomography. Diabetes 44(1):31–36PubMed
6.
Donahue RP, Bean JA, Donahue RA, Goldberg RB, Prineas RJ (1997) Insulin response in a triethnic population: effects of sex, ethnic origin, and body fat. Miami Community Health Study. Diabetes Care 20(11):1670–1676PubMed
7.
Kuhl J, Hilding A, Ostenson CG, Grill V, Efendic S, Bavenholm P (2005) Characterisation of subjects with early abnormalities of glucose tolerance in the Stockholm Diabetes Prevention Programme: the impact of sex and type 2 diabetes heredity. Diabetologia 48(1):35–40PubMed
8.
Macotela Y, Boucher J, Tran TT, Kahn CR (2009) Sex and depot differences in adipocyte insulin sensitivity and glucose metabolism. Diabetes 58(4):803–812PubMed
9.
Toth MJ, Tchernof A, Sites CK, Poehlman ET (2000) Effect of menopausal status on body composition and abdominal fat distribution. Int J Obes Relat Metab Disord 24(2):226–231PubMed
10.
Sites CK, Toth MJ, Cushman M, L’Hommedieu GD, Tchernof A, Tracy RP, Poehlman ET (2002) Menopause-related differences in inflammation markers and their relationship to body fat distribution and insulin-stimulated glucose disposal. Fertil Steril 77(1):128–135PubMed
11.
Svendsen OL, Hassager C, Christiansen C (1995) Age- and menopause-associated variations in body composition and fat distribution in healthy women as measured by dual-energy X-ray absorptiometry. Metabolism 44(3):369–373PubMed
12.
Brochu M, Starling RD, Tchernof A, Matthews DE, Garcia-Rubi E, Poehlman ET (2000) Visceral adipose tissue is an independent correlate of glucose disposal in older obese postmenopausal women. J Clin Endocrinol Metab 85(7):2378–2384PubMed
Related posts:
 Influence of Ovarian Hormones on Skeletal Muscle Contractility
Influence of Ovarian Hormones on Skeletal Muscle Contractility
 Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
Transitions Across a Lifetime: Unique Cardiovascular Physiology of Women and Relationship to Cardiovascular Disease Risk
 Metabolic Health in the Aging Female: Human Perspective
Metabolic Health in the Aging Female: Human Perspective
 Estrogen Effects on Skeletal Muscle
Estrogen Effects on Skeletal Muscle
 Novel Findings in Bone Biology: Impact on Bone Health for Women
Novel Findings in Bone Biology: Impact on Bone Health for Women
 The Impact of Estrogen Receptor α Expression in the Pathogenesis of the Metabolic Syndrome
The Impact of Estrogen Receptor α Expression in the Pathogenesis of the Metabolic Syndrome
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


