Proinflammatory
Anti-inflammatory
Omega-6 fatty acids
Omega-3 fatty acids
Saturated fatty acids
Polyphenols
Excess carbohydrates
The body requires a balance of these proinflammatory and anti-inflammatory nutrients in the diet to maintain a balance in the initiation and resolution phases of the inflammatory process. However, an imbalance in either an excess of proinflammatory nutrients or deficiency of anti-inflammatory nutrients will increase the activity of NF-κB.
Once NF-κB is activated, it travels from the cytoplasm into the nucleus of the cell to cause the transcription of a wide variety of inflammatory proteins such as proinflammatory cytokines (IL-1β, IL-6, TNF-α, and others) as well as the increased production of the COX-2 enzyme which is needed for the production of proinflammatory eicosanoids such as prostaglandins. These inflammatory mediators expressed as a result of NF-κB activation act as paracrine hormones to activate nearby cells via their cytokine or prostaglandin receptors to amplify their inflammatory responses.
How these dietary factors impact both the initiation and resolution of inflammation at the molecular level is described in the next section.
1.3 Proinflammatory Nutrients
Foremost of the proinflammatory nutrients are omega-6 fatty acids as these are the building blocks required for the formation of hormones known as eicosanoids that are key players in the inflammatory process. Although there are hundreds of known eicosanoids, the primary eicosanoids important for the initiation phase of inflammation are prostaglandins and leukotrienes. In particular, the most common dietary omega-6 fatty acid, linoleic acid, must be first converted into arachidonic acid (AA) that is the building block for prostaglandins and leukotrienes. The synthesis of AA is tightly controlled by two distinct enzymes, delta-6 and delta-5 desaturase.
These two particular rate-limiting enzymes are controlled by the hormones insulin and glucagon that are generated by the balance of carbohydrates and protein in the diet [6]. It is the balance of these carbohydrates and protein at each meal that influences the secretion of insulin (an activator for both desaturase enzymes) and glucagon (an inhibitor of both desaturase enzymes). The dietary levels of long-chain omega-3 fatty acids (eicosapentaenoic acid (EPA) and docosahexaenoic acid (DHA)) are also important as they can be weak feedback inhibitors of the activity of both desaturase enzymes [6]. However, under conditions of insulin resistance, the levels of insulin in the blood will increase often causing an overproduction of AA because of the activation of both desaturase enzymes. This increases the potential for the generation of proinflammatory prostaglandins and leukotrienes derived from AA.
Linoleic acid is also very prone to oxidation thus increasing the formation of increased oxidative stress within the cell. Oxidative stress is another activator of NF-κB [7]. This is especially true if the diet is low in polyphenols that can function as powerful antioxidants to retard the oxidation of linoleic acid [8]. Finally, as the levels of omega-6 fatty acids increase in the diet, the endogenous production of anti-inflammatory omega-3 fatty acids (EPA and DHA) is decreased [9].
Saturated fatty acids are not as proinflammatory as omega-6 fatty acids, but they still have the potential to activate NF-κB. This is done by binding to toll-like receptors (TLR), especially TLR-4 [10]. Once TLR-4 is activated, through a series of signaling mechanisms, it can also increase the activation of NF-κB [11]. Saturated fatty acids also can increase the formation of lipid rafts in the cell membrane that amplifies the signals coming from TLR-2 and TLR-4 to further increase NF-κB activation [12].
The body (in particular the brain) needs a certain level of glucose for optimal function. Similar to linoleic acid, glucose is also prone to oxidation and can form advanced glycosylated end products (AGE), which are glycosylated proteins that can interact with their receptors (RAGE) on the cell surface that likewise activate NF-κB [13]. Excess intake of carbohydrates (especially those containing high levels of glucose such as grains and pasta) will increase the secretion of insulin by the pancreas to bring the elevated levels of blood glucose back within a normal operating range. However, with the development of insulin resistance, insulin levels remain continuously elevated increasing the likelihood of more dietary linoleic acid becoming transformed into AA.
1.4 Anti-inflammatory Nutrients
Just as there are proinflammatory nutrients that activate NF-κB, there is a wide range of nutrients that decrease its activity. Thus an anti-inflammatory nutrient is defined as one that ultimately inhibits activation of NF-κB.
The anti-inflammatory potential of EPA and DHA is manifold. As pointed out above, they are weak inhibitors of the delta-5 and delta-6 desaturase enzymes that will thus decrease the production of AA [6, 9]. Any decrease in AA will restrict the production of proinflammatory eicosanoids. They also compete with AA as substrates for the lipo-oxygenase (LOX) enzymes and cyclooxygenase (COX) enzymes required to make leukotrienes and prostaglandins. However, the bulky three-dimensional structure of DHA makes it a poor substrate for the COX enzymes. Since the COX-2 enzyme is one of those proteins that have increased expression by the activation of NF-κB, this suggests that EPA may be the more anti-inflammatory of the two omega-3 fatty acids to reduce the impact of diet-induced inflammation.
Omega-3 fatty acids also can be sensed by nutrient receptors such as GPR120 on the cell surface to signal the inactivation of NF-κB [14, 15].
Polyphenols also represent anti-inflammatory nutrients [16]. Although not as strong regulators of resolution as are the omega-3 fatty acids, they do act on different aspects of the initiation of inflammation and therefore are complimentary to omega-3 fatty acids.
The first of these polyphenol pathways is their action as antioxidants [16, 17]. Increased oxidation within the cell can increase NF-κB activation [7]. One of the primary sources of diet-induced inflammation is increased consumption of calories [4, 18]. Any excess calories that cannot be immediately converted into chemical energy are typically stored for future use. This conversion of excess calories into new molecules for long-term storage will generate excessive levels of free radicals that are the underlying cause of oxidative stress. As powerful antioxidants, polyphenols are key to preventing the excess buildup of free radicals. Furthermore, polyphenols can interact with other gene transcription factors, such as Nrf2, to cause the increased production of additional antioxidative enzymes such as superoxide dismutase and glutathione peroxidase that will further reduce oxidative stress [16, 17, 19].
At higher levels of dietary intake, polyphenols (as well as omega-3 fatty acids) can activate the anti-inflammatory gene transcription factor PPARγ which not only inactivates NF-κB but also increases the production of new healthy fat cells needed to prevent the lipotoxicity of stored fat from spreading into other tissues such as the liver and muscles that can increase insulin resistance in those organs [20, 21].
At still higher levels of dietary intake, polyphenols can activate the gene transcription factor SIRT-1 that causes the increased production of AMP kinase (AMPK) that acts as the controller of metabolism [22, 23]. With the activation of AMPK, catabolism and autophagy are activated to increase ATP levels and the anabolism is decreased [23].
1.5 Pro-resolution Nutrients
The definition of a pro-resolution nutrient is one that promotes the resolution of inflammation. This is a very different concept than simply the inhibition of inflammation. The initiation phase of inflammation will continue unless bought back to equilibrium by the resolution phase of inflammation. Increased levels of neutrophils at the injury site characterize the acute initiation of inflammation [5, 24]. On the other hand, increased levels of macrophages at the same site characterize the potential beginning of the resolution phase [5, 24]. However, without adequate levels of resolvins, these macrophages at the injury site are maintained in the activated M1 state and remain as potent generators of proinflammatory cytokines [25]. It is the resolvins that cause the reversion of the activated, proinflammatory macrophages in the M1 state to become anti-inflammatory M2 state macrophages (Fig. 1.1). Without this macrophage transition mediated by resolvins, the inflammatory response continues at a lower but still proinflammatory level. This constitutes cellular inflammation.
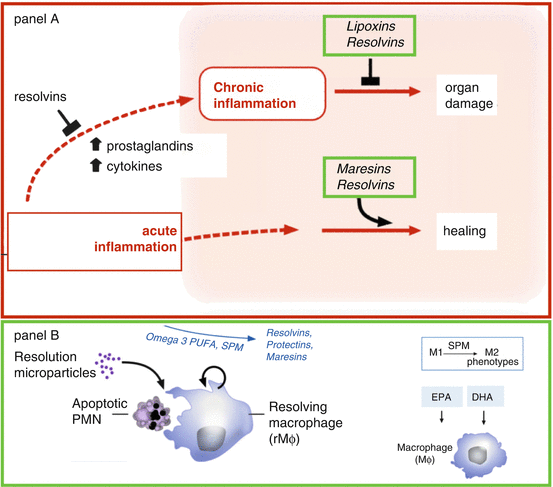
Fig. 1.1
PANEL (A) Specialized pro-resolving mediators (SPM) such as lipoxins, resolvins, protectins and maresins stimulate cellular events that counter-regulate pro-inflammatory mediators and regulate PMN, monocyte and macrophage response, leading to resolution. PANLE (B) Nutritional abundance of Omega-3, PUFA yields precursors of specialized pro-resolving mediators of inflammation, and eventually the M1 to M2 macrophage turnaround. Modified after Serhan [25]
The combination of adequate dietary intake of long-chain omega-3 fatty acids and polyphenols coupled with the dietary restriction of omega-6 and saturated fats as well as excess carbohydrates and calories represents the best therapeutic approach to maintain a balanced inflammatory response of initiation and resolution.
1.6 Adipose Tissue and Inflammation
The most effective site for storage of excess fat calories is the adipose tissue including those excess calories from carbohydrates that are converted to fat in the liver [26]. The fat cells of the adipose tissue are the only cells in the body that are designed to safely contain large amounts of fat. This is why the adipose tissue is extremely rich in stem cells that can be converted to new fat cells to contain large levels of excess energy as triglycerides [27]. As long as those fat cells are healthy, there are no adverse metabolic effects (except excess weight) for the person. This is why approximately one-third of obese individuals fall into the category of “metabolically healthy obese” [28]. They have excess body fat but no metabolic disturbances that characterize the manifestation of insulin resistance.
However, fat cells do not have an unlimited capacity to expand. Even though the adipose tissue is highly vascularized, the overexpansion of existing fat cells can create hypoxia that activates the HIF-1 gene [29, 30]. This results in the increased expression of both JNK and IKK, thereby creating inflammation within the fat cell [31]. This inflammation in turn creates insulin resistance within the fat cell.
In the adipose tissue, insulin is normally an anti-lipolytic hormone as it decreases the activity of the hormone-sensitive lipase (HSL) that is required to release stored fatty acids [32]. With the development of cellular inflammation and insulin resistance in the fat cell, higher levels of FFA can leave the fat cell to enter into the circulation to be taken up by other organs such as the liver and the skeletal muscles that are unable to safely store large amounts of fat. As described later, this leads to developing insulin resistance in these organs. With increased inflammation in the fat cells, there also is a migration of greater numbers of M1 macrophages into the adipose tissue with a corresponding release of inflammatory cytokines such as TNF-α and a family of proinflammatory cytokines that further increases insulin resistance and lipolysis [33] (Fig. 1.2). In the lean individual, only about 10 % of the adipose tissue mass is composed of macrophages, and those macrophages are primarily in the anti-inflammatory M2 state [34]. In the obese individual, up to 50 % of the mass of the adipose tissue may contain macrophages but now in the activated proinflammatory M1 state [34]. Theoretically, new healthy fat cells could be generated from stem cells within the adipose tissue. However, that process requires the activation of the gene transcription factor PPARγ [35]. The activity of this gene transcription factor is inhibited by inflammatory cytokines such as TNF-α [36]. On the other hand, the activity of PPARγ is increased in the presence of anti-inflammatory nutrients such as omega-3 fatty acids and polyphenols [37]. Without the ability to form new healthy fat cells, the continued expansion of the existing fat cells eventually leads to cell death and further adipose tissue inflammation caused by incoming neutrophils and macrophages to clean the cellular debris caused by the necrotic fat cells [38].
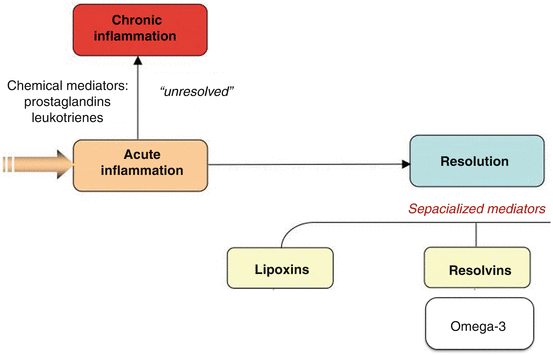
Fig. 1.2
Chronic inflammation can result from unresolved inflammatory responses. Arachidonic-acid-derived lipid mediators such as proinflammatory prostaglandins and leukotrienes can amplify this process. Bioactive products of omega-3 were originally isolated from mouse resolving exudates. The structure of these products was elucidated, and the biosynthesis of each new omega-3 family from EPA and DHA was recapitulated and analyzed in humans
As stated earlier, insulin resistance can inhibit the action of hormone-sensitive lipase (HSL) leading to the elevation of insulin levels in the blood due to systemic insulin resistance in the muscle cells. Ironically, the increased hyperinsulinemia activates the lipoprotein lipase at the surface of the fat cell that hydrolyzes lipoprotein triglycerides to release free fatty acids [39] as well as increases the synthesis of fatty acid-binding proteins that bring the newly released FFA from the lipoproteins to the fat cells for deposition [40]. The increase in fatty acid flux into the fat cells also requires greater synthesis of the FFA into triglycerides, but this can lead to ER stress activating the JNK pathway thus further increasing insulin resistance in the fat cells [41]. This sets up a vicious cycle in which insulin resistance results in greater hunger (via insulin resistance in the hypothalamus) with increasing flux of FFA both into and out of the adipose tissue [42]. The cytokines being released by the proinflammatory M1 macrophages being attracted to the adipose tissue due to increasing cellular inflammation only increase this process by accelerating insulin resistance in the fat cells. This is why obese individuals with insulin resistance have greater levels of both the uptake and release of FFA into and from the adipose tissue. The increase in lipid influx causes an overload of the synthetic capacity to make triglycerides, and as a result, both DAG and ceramide levels begin to increase which only further increases insulin resistance in the fat cells [43].
The speed of the inflammatory changes in the adipose tissue is not as rapid as they are in the hypothalamus. Whereas inflammatory changes can be seen in the hypothalamus within 24 h after beginning an HFD, it often takes 12–14 weeks to see similar changes in inflammation in the adipose tissue [44].
If the fat cells cannot expand rapidly enough to store this increasing fatty acid flow, then the excess released fatty acids begin to accumulate in other tissues such as the liver and skeletal muscles, and this begins the process of lipotoxicity that further increases systemic insulin resistance [45]. It is with the development of lipotoxicity that the real metabolic consequences of insulin resistance begin.
We often think of obesity as the only cause of insulin resistance, yet the genesis of insulin resistance appears to start in the hypothalamus with a disruption in the normal balance of hunger and satiety signals. As hunger increases, so does calorie intake.
1.7 Hypothalamus
In many ways, early commands to develop insulin resistance in the body appear to start in the hypothalamus. The hypothalamus acts to match energy intake to energy expenditure to prevent excess accumulation of stored energy [46]. In particular, satiety signals from the gut are matched to adiposity (primarily leptin) and blood (primarily insulin) hormonal signals to control food intake [47]. Unfortunately, either excess calories or saturated fats (especially palmitic acid) can cause inflammation in the hypothalamus leading to resistance to the satiety signaling of both insulin and leptin [48, 49]. As a result, satiety is attenuated and hunger increases. Since the hypothalamus also contains GPR120 binding proteins, the presence of adequate levels of omega-3 fatty acids in this tissue can also decrease inflammation within the hypothalamus [50]. In fact, intracerebroventricular (icv) injections of omega-3 fatty acids into obese rats decrease insulin resistance [51]. Likewise similar icv injections of anti-TLR-4 and anti-TNF-α antibodies also decrease insulin resistance [52].
High-fat diet (HFD), especially those rich in saturated fats, is the standard method to cause diet-induced obesity. Increased inflammation appears in the hypothalamus within 24 h after beginning an HFD as indicated by increases in JNK and IKK proteins as well as increased expression of TLR-4 receptors and detection of ER stress [53]. IKK induces inflammation via activation of NF-κB that inhibits the normal hormonal signaling of leptin and insulin that is necessary to create satiety. Activation of JNK is often preceded by the increase in ER stress [54]. This sets up a vicious cycle of increased hunger that eventually leads to the accumulation of excess calories as stored fat in the adipose tissue. It should be noted that the inflammation in the hypothalamus precedes any weight gain in the adipose tissue [55]. This also explains why significant calorie restriction can reduce insulin resistance before any significant loss in excess body fat in the adipose tissue. These experimental observations suggest that the hypothalamus is the central control point for the development of insulin resistance.
Excess nutrient intake (especially saturated fat) can also indirectly cause inflammation in the hypothalamus by activation of the TLR-4 receptors in the microglia in the brain eventually causing inflammatory damage to neurons in the hypothalamus [49]. It has been shown that with an extended use of an HFD, there is a decrease in the number of neurons responsible for generating satiety signals in the hypothalamus [56].
HFD diets are also associated with increased production of palmitic acid-enriched ceramides in the hypothalamus which would provide still another link to the increased insulin and leptin resistance that gives rise to increased hunger as satiety depends on functioning insulin pathways in the hypothalamic neurons [57].
Besides the presence of the GPR120 receptors in the hypothalamus, which if activated by omega-3 fatty acids decreases inflammation, there are other fatty acid nutrient sensors in the hypothalamus that can be activated to increase inflammation. In particular, any increase in the free fatty acid (FFA) levels in the blood can be sensed by CD36/FATP-1 transporter at the surface of blood-brain-barrier (BBB). If those fatty acids are rich in palmitic acid (the primary product of de novo lipid production in the liver caused by excess dietary glucose), then the HPA axis is activated to release more cortisol that increases insulin resistance [58]. On the other hand, if the fatty acid being sensed is primarily oleic acid, there will be a reduction in NPY (a powerful appetite-inducing hormone) expression in the hypothalamus that promotes satiety [59].
Finally there is the interaction of the hypothalamus with the liver via signaling through the vagus nerve [53]. This may explain why any inhibition of TNF-α or TLR-4 signaling in hypothalamus also decreases glucose production in the liver [60, 61].
As you can begin to appreciate, the central regulation of appetite control by the hypothalamus is a very complex orchestration of the levels of inflammation and nutrient intake generated by the diet and the sensing of those levels by the hypothalamus.
1.8 Insulin Resistance
Although the definition of insulin resistance is deceptively simple (a condition in which cells are no longer responding appropriately to circulating insulin), the molecular causes of insulin resistance are diverse and extremely complex.
It is known that certain short-term dietary changes can rapidly reduce insulin resistance before any significant fat loss occurs. This would include strict calorie restriction that can reduce insulin resistance within a matter of days [62]. Likewise certain drugs such as corticosteroids can rapidly increase insulin resistance [63].
Furthermore, there are various metabolic adaptations to stressors that can induce insulin resistance. These stressors include pregnancy, hibernation, and sepsis [1]. The increase in insulin resistance in response to these stressors is a method of diverting stored nutrients to address the necessary metabolic adaptation. Likewise sleep deprivation is another effective way of increasing insulin resistance in the short-term [64].
However, the chronic insulin resistance appears to be directly or indirectly related to diet-induced inflammation. The mechanisms at the molecular level are complex and manifold, but they are based on the ability of increased cellular inflammation to interrupt insulin’s action by disruption of the signaling mechanisms within the cell. The primary suspects appear to be the inflammatory cytokine tumor necrosis factor alpha (TNF-α) and other inflammatory protein kinases such as c-Jun N-terminal kinase (JNK) and the inhibitor of IκKβ kinase (IKK) [65].
TNF-α knockout models are resistant to the development of insulin resistance in animal strains prone to diet-induced obesity (DIO mice) or those that lack leptin (Ob/Ob mice) [31, 66]. Activation of JNK and IKK interrupts insulin-signaling pathways via separate mechanisms. Activation of IKK causes the dissociation of the inhibitory protein that prevents the translocation of NF-κB into the nucleus [67]. Once IKK releases that inhibitory protein from NF-κB, that gene transcription can now move to the nucleus to cause the expression of inflammatory proteins. The JNK pathway is stress activated and is associated with the presence of M1-activated macrophages [41].
The realization that inflammation was related to insulin resistance was more than century ago when it was observed that certain anti-inflammatory drugs (salicylates and aspirin) were effective in reducing the hyperglycemia observed in diabetes [68, 69]. It is now known that these drugs are inhibitors of IKK [70].
Additional molecular mechanisms of insulin resistance include the lipid overload hypothesis in which there is a buildup of diacylglycerides (DAG) or ceramides that inhibit the signaling of insulin as well as endoplasmic reticulum (ER) stress (induced by excess calories) or oxidative stress (induced by the generation of excess free radicals) [39–41, 71]. Making these diverse molecular mechanisms even more complex is that they are operative in some organs and not in others.
1.9 Individual Organ Responses to Diet-Induced Inflammation and Insulin Resistance
1.9.1 Liver
The liver can be viewed as the central manufacturing plant in the body. Raw materials (primarily carbohydrates and fats) are bought into the body to be processed by the liver and either stored (as liver glycogen) or repackaged as newly formed triglycerides (in the form of lipoproteins). The liver helps maintain stable glucose levels between meals by balancing glycogenesis (glycogen formation) and glycolysis of stored glycogen [72]. It should be pointed out that the glycogen stored in muscles can only be used internally as a source of energy and can’t be released back into the circulation to help maintain stable blood glucose levels.
Unlike the adipose tissue that can safely store excess fat, the liver cannot. Therefore of the first adverse metabolic consequences of insulin resistance is the buildup of fatty deposits in the liver. This is known as nonalcoholic fatty liver disease or NAFLD. Currently 20–30 % of Americans have NAFLD and 90 % of obese type 2 diabetic patients have NAFLD [73]. Ominously, it is estimated that 50 % of all Americans will have NAFLD by 2030 [72].
Another difference between the liver and the adipose tissue is the lack of infiltrating macrophages. Whereas a significant increase is observed in the levels of macrophages in the adipose tissue upon inflammation, it is the internal macrophages (Kupffer cells) in the liver that become activated. These activated Kupffer cells can now release cytokines that will further activate NF-κB in the liver cells.
Like hypothalamic inflammation, NAFLD can be rapidly generated in animal models within 3 days of starting an HFD [74]. This may be due to the direct linkage of the hypothalamus to the liver via the vagus nerve [75]. Once NAFLD is established, the ability of insulin to suppress liver glucose production is diminished without changes in weight, fat mass, or the appearance of any indication of insulin resistance in the skeletal muscle [76].
Because of the rapid buildup of fatty acids in the liver, the ability to convert them to triglycerides is also overwhelmed and DAG formation in liver increases [77, 78]. This is why the levels of DAG in the liver are the best clinical marker that chronic insulin resistance has begun to develop in that organ. The primary source of the fatty acids coming to the liver is via the adipose tissue because as the adipose tissue develops insulin resistance, the increased flow of FFA from the fat cells into the blood and therefore into the liver increases [79]. De novo lipid synthesis of fats from glucose in the liver is a smaller contributor to this increased flux of FFA into the liver [80]. Furthermore, liver insulin resistance is related only to the fatty acid levels in the liver, not the levels of visceral fat [81]. This may explain why many normal-BMI individuals (especially Asians) can have high levels of insulin resistance in the liver [82].
Since the liver also controls cholesterol synthesis, insulin resistance in this organ is reflected in growing dysfunction in lipoprotein synthesis. In particular VLDL particles are increased and HDL levels are decreased [72]. This is easily measured by the TG/HDL ratio that is a good general clinical marker for liver insulin resistance [83].
1.9.2 Skeletal Muscle
Skeletal muscle represents the key site for glucose uptake. Thus reducing insulin resistance in this organ becomes a primary strategy for managing diabetes. Unlike the adipose tissue, where macrophage infiltration is a key indicator of inflammation, there is very little macrophage infiltration observed in the skeletal muscle in individuals with insulin resistance [84]. It appears that cytokines coming from other organs (adipose tissue and liver) may have the important impact on the development of insulin resistance in the muscle. However, enhanced signaling through the TLR-4 receptor by saturated fatty acids can reduce fatty acid oxidation of the lipids in the muscle [85]. In addition, palmitic acid is the preferred substrate for ceramide synthesis [86]. Whereas ceramide levels are not related to insulin resistance in the liver, they are strongly related to insulin resistance in the muscle [87]. This suggests that the molecular drivers of insulin resistance can be different from organ to organ.
1.9.3 Pancreas
Although the beta cells of the pancreas sense glucose levels in the blood and secrete insulin in response to those levels, the beta cells of this organ are not normally considered targets of insulin resistance. However, the beta cells are very prone to toxicity mediated by inflammatory agents. In particular, 12-HETE, derived from AA, is very toxic to the beta cells [88]. With the destruction of the beta cells by 12-HETE, the pancreas is no longer able to maintain compensatory levels of insulin secretion to reduce blood glucose levels, and the development of type 2 diabetes is rapid.
Related posts:
 Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
Nutrition Around the Clock and Inflammation: The Right Food at the Right Time Makes the Difference
 Obesity and Inflammation in Pregnancy
Obesity and Inflammation in Pregnancy
 PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
PCOS and Pregnancy: Impact of Endocrine and Metabolic Factors
 The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development
The Role of Placental Inflammasomes in Linking the Adverse Effects of Maternal Obesity on Fetal Development
 Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
Gastrointestinal Symptoms and Nutritional Profile During Pregnancy
 Maternal Diet, Developmental Origins, and the Intergenerational Transmission of Cardiometabolic Traits: A Window of Opportunity for the Prevention of Metabolic Syndrome?
Maternal Diet, Developmental Origins, and the Intergenerational Transmission of Cardiometabolic Traits: A Window of Opportunity for the Prevention of Metabolic Syndrome?
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


