FIGURE 3.1
3.1 HyperTG NormoapoB
This can be due to (Figure 3.1):
1)
Increased chylomicron particles
2)
Increased chylomicron particles and increased VLDL particles
3)
Increased chylomicron and VLDL remnants particles
4)
Increased VLDL particles
3.1.1 HyperTG NormoapoB due to Increased Chylomicron Particles
3.1.1.1 Diagnosis
Hyperchylomicronemia is present in patients with marked hypertriglyceridemia (triglycerides > 6 mmol/l) in combination with an apoB < 0.75 g/l and a TG/apoB ≥ 10.90-94The combination of very high triglycerides and low apoB is the key to diagnosis.
3.1.1.2 Pathophysiology
Chylomicrons are the largest, most triglyceride-rich apoB particles and are synthesized and secreted by the intestine. The triglyceride within chylomicrons is normally rapidly hydrolyzed by lipoprotein lipase (LPL) in adipose tissue and skeletal muscle and the remnant particle that is produced, which contains some of the original triglyceride and all of the original cholesterol, is then rapidly removed by the liver (Figure 3.3 and see Figure 1.2 ). Because each chylomicron particle contains so much triglyceride – a chylomicron particle can reach 1200 nm or 8000 angstroms in size (see Table 1.1) – it requires only a relatively small number of chylomicron particles to produce very elevated triglyceride levels.
FIGURE 3.2
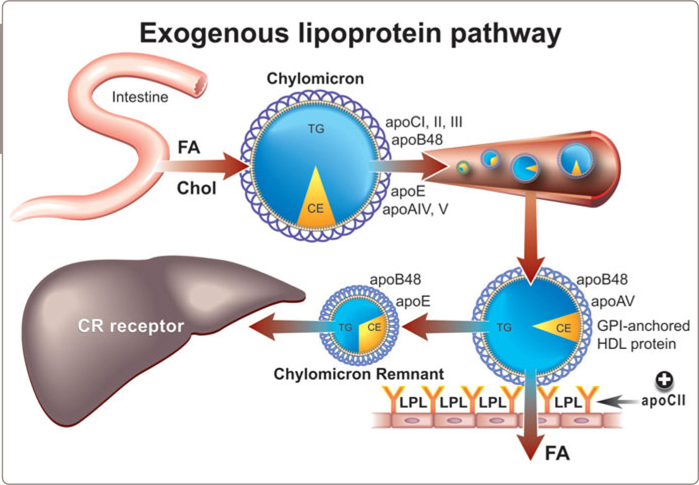
FIGURE 3.3
In hyperchylomicronemia, the total apoB is strikingly low. The low total apoB is predominantly the effect of a low LDL apoB. The LDL particles appear to be enriched in triglyceride and depleted in cholesterol (Table 2.2 ); HDL-C is low: all findings, which might be anticipated given the high plasma triglyceride levels and therefore the greatly increased mass of triglyceride available for cholesterol ester transfer protein (CETP)-mediated core lipid exchange (Figure 1.12 ).
3.1.1.3 Causes
The principal genetic cause of marked hyperchylomicronemia is failure to synthesize active LPL (Figure 3.4 ).90,92 Most cases are compound heterozygotes due to mutations in exons 5 and 6 of the LPL gene and involve either the catalytic or the heparin-binding domain. A mutation in apoCII, an essential co-factor for LPL activity, is a less common but also well-established cause of this syndrome. A few cases have been related to a defect in glycophosphatidylinositol (GPI −) anchored HDL binding protein, a protein, whose function is to bind LPL to the endothelium95 and there is considerable interest in the critical role that apoAV may play in triglyceride clearance from plasma (Figure 3.5 ).96
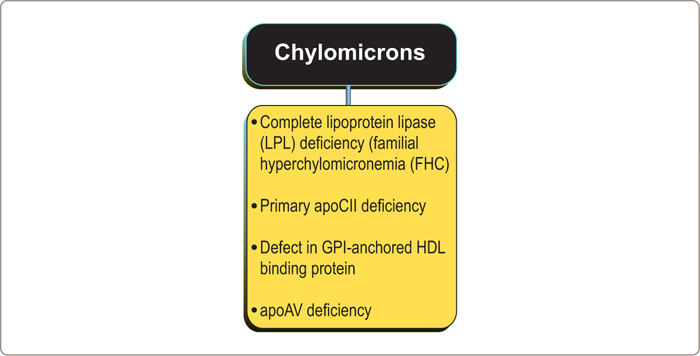
FIGURE 3.4
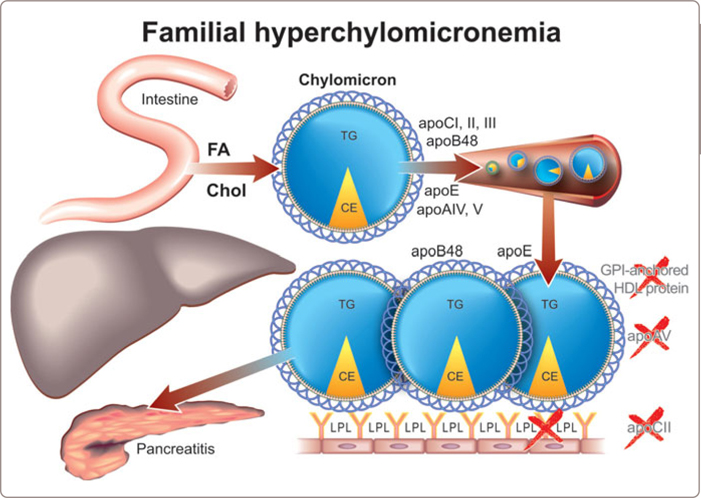
FIGURE 3.5
3.1.1.4 Clinical Diagnosis
Hyperchylomicronemia is rare: approximately 1 per 1 million in the population. Chylomicrons are too large to penetrate the vascular wall with any facility and so vascular disease is not a problem. Rather, pancreatitis is the clinical risk from severe hyperchylomicronemia with microvascular obstruction due to the chylomicron particles a popular hypothesis. The risk becomes substantial when plasma triglyceride levels are more than 10 mmol/l. However, some patients will not develop pancreatitis even with triglyceride levels as high as 100 mmol/l whereas others suffer acute pancreatitis at triglyceride levels of 5 mmol/l.
A major clinical feature is eruptive xanthomata (Figure 3.6 ), small, yellowish-white papules that often appear in clusters, on the back, buttocks, and extensor surfaces of the arms and legs. Lipemia retinalis is another major clinical feature – the retinal blood vessels on fundoscopic examination are opalescent as shown in Figure 3.7 which disappears after treatment of the hypertriglyceredimia (Figure 3.8 ). Finally, hepatosplenomegaly due to the uptake of circulating chylomicrons by reticuloendothelial cells in the liver and spleen can be found on physical examination. A blood sample may appear pale and the serum milky (Figure 3.9 ). Pseudohyponatremia may be present but this is less likely given newer laboratory methods. Occasionally plasma must be diluted in order to recognize the true extent of the elevated triglycerides.
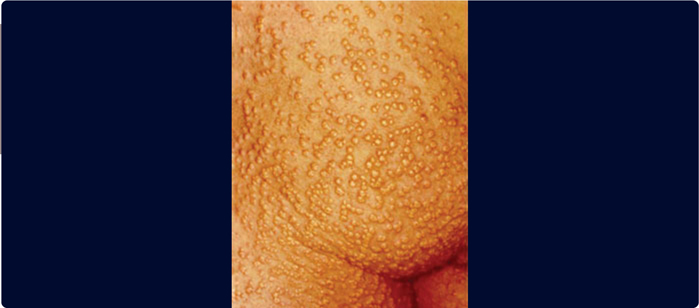
FIGURE 3.6
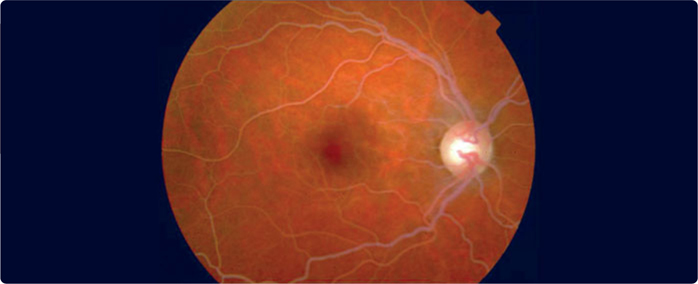
FIGURE 3.7
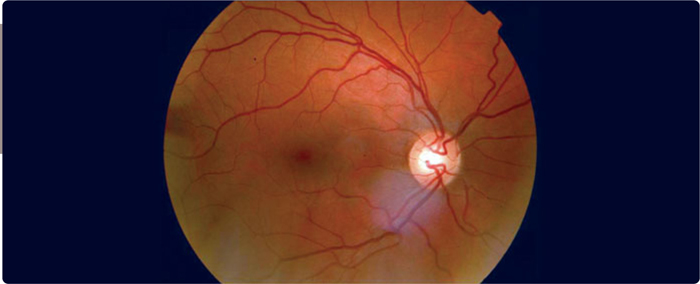
FIGURE 3.8
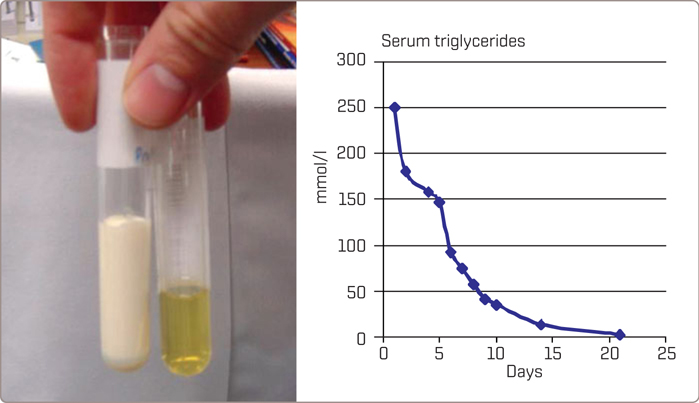
FIGURE 3.9
Since triglyceride levels tend to decrease substantially in the first 48 hours after the onset of pancreatitis, delay in presentation may obscure diagnosis of this as the cause of the acute pancreatitis. Large increases in serum amylase may not be present and an elevated pancreatic lipase may, in fact, be a more reliable indicator of the problem.92Imaging with MRI or CT that demonstrates intrapancreatic or peripancreatic edema is the most reliable tool to diagnose the pancreatitis. The initial attack of acute pancreatitis may result in pseudocysts, which increase the potential for further clinical episodes.
3.1.1.5 Treatment
Fasting for at least 24 hours is the first priority in treatment for patients with acute pancreatitis due to hyperchylomicronemia. When there is no dietary intake of cholesterol and triglycerides, no chylomicrons will be formed, and triglycerides will fall to within the normal range within days (Figure 3.9 ) Intravenous fluids are administered as necessary and plasmapheresis can be considered if available and if the patient is critically ill. An insulin drip may be effective to improve activity of LPL even when blood glucose is not elevated. For patients not admitted to hospital, the major therapeutic intervention in familial chylomicronemia syndromes is dietary fat restriction (to as little as 15 g/day to limit the formation of chylomicrons) with fat-soluble vitamin supplementation.
Long-term management of these patients involves lifestyle changes (avoidance of alcohol, weight reduction and low-fat diet). The value of medium-chain triglycerides needs to be better defined as clinical reports of their efficacy differ.97 Medications are often considered in patients with fasting plasma triglyceride levels > 10 mmol/l with a target of triglycerides < 5 mmol/l. Unfortunately, medications such as fibrates generally do little. Management of patients with familial chylomicronemia syndrome is particularly challenging during pregnancy and may require plasmapheresis to remove the circulating chylomicrons (see also Table 4.5 Chapter 4 ). The EVOLVE (Epanova for Lowering Very High Triglycerides) study reported success with the free fatty acid (FA) form of omega-e FA in severe hypertriglyceridemia.98 Of considerable interest is a report documenting marked reductions in plasma triglycerides in 3 patients who were either homozygotes or compound heterozygotes for LPL deficiency with infusion of an inhibitor of apoCIII messenger RNA.99 Given that apoCIII is thought to inhibit LPL activity, which is genetically absent in most of these patients, these positive results are welcome, but surprising, and indicate how much remains to be learned about triglyceride clearance from plasma. Work is underway to develop a gene therapy approach100and at least acute therapy with a microsomal triglyceride transport inhibitor such as lomitapide might be considered.101
3.1.2 HyperTG NormoapoB due to Elevated Chylomicrons and VLDL Particles
3.1.2.1 Diagnosis
These patients have marked or severe hypertriglyceridemia (triglycerides > 6 or 10 mmol/l) due to increased numbers of chylomicrons and VLDL particles (Figure 3.10 ). The triglyceride-rich particles account for a triglyceride/apoB ratio ≥ 10 and the syndrome is distinguished from pure hyperchylomicronemia by an apoB > 0.75 g/l. Most of these patients have a normal apoB but the occasional patient has an elevated value due to an associated increase in LDL particle number (see Table 2.1 ).
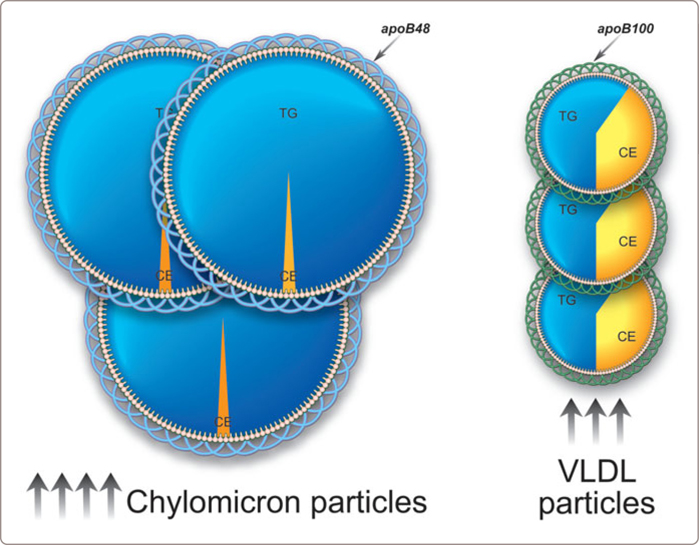
FIGURE 3.10
3.1.2.2 Pathophysiology
As well as chylomicrons, LPL hydrolyzes triglycerides in VLDL particles, which is the first step in their conversion to LDL particles. An increase in VLDL production is thought to saturate triglyceride clearance. If this occurs, even modest increases in production will produce profound increases in plasma triglycerides.
3.1.2.3 Causes
There are no known primary causes for this disorder but it can be associated with a partial LPL deficiency or an abnormality in apoAV, an apolipoprotein, which appears to be involved in the association of LPL with triglyceride-rich particles (Figure 3.11 ).

FIGURE 3.11
However, when partial LPL or apoAV deficiency is associated with a secondary factor, such as abdominal obesity, type 2 diabetes mellitus (DM2), alcohol abuse, or medications such as steroids or estrogens or isotretinoin (Accutane), then severe hypertriglyceridemia due to accumulation of chylomicron and VLDL particles may result. Pregnancy92 can also provoke the clinical expression of this phenotype. Beta-blockers and thiazide therapy have also been implicated. If beta-blocker therapy is required, the metabolic side effect profile of agents such as carvedilol is preferable to those such as metoprolol. In these patients, triglyceride clearance is already severely compromised. Therefore, relatively modest increases in VLDL production can produce profound increases in plasma triglyceride levels.
3.1.2.4 Clinical Presentations
Patients with severe hypertriglyceridemia due to chylomicrons and VLDL particles may present with eruptive xanthomata and plasma may appear milky (see Figures 3.6 , 3.7 , 3.8 and 3.9 ). If chylomicron and VLDL particle numbers are increased sufficiently, pancreatitis may occur. Plasma apoB is only occasionally elevated above the 75th percentile (i.e. > 1.2 g/l).
3.1.2.5 Treatment
In patients with severe hypertriglyceridemia who are at risk of pancreatitis, dietary total fat intake should be reduced acutely. Secondary causes should be identified and, if present, treated. Treatment with fibrates or omega-3 FA may be helpful in these patients, particularly when plasma triglycerides have fallen due to restriction of fat intake if partial LPL activity is present. Nicotinic acid is also an option. If the patient is diabetic, an insulin drip may be helpful. Whether LDL-lowering therapy with statins is indicated depends on the level of apoB and the clinical circumstances. Patients with increased cardiovascular risk because of the presence of DM2 or a history of cardiovascular disease (CVD) will require LDL-lowering therapy to reduce apoB levels to at least < 0.75 g/l and, if appropriate to < 0.65 g/l (see Chapter 6 ). The same range of options is available to treat the severe hypertriglyceridemia in these patients as in those with hyperchylomicronemia (see 3.1.1 and Table 4.3 Chapter 4 ).
3.1.3 HyperTG NormoapoB due to Remnant Lipoprotein Disorder
3.1.3.1 Diagnosis
In patients with combined hypercholesterolemia and hypertriglyceridemia and normal apoB, one should always consider whether remnant lipoprotein disorder is present. The diagnostic criteria in the apoB algorithm are: apoB < 1.20 g/l, TG ≥ 1.5 mmol/l, triglyceride/apoB < 10, total cholesterol/apoB ≥ 6.2. Characteristic for remnant lipoprotein disorder is the accumulation of chylomicron and VLDL remnant particles (Figure 3.12 ).
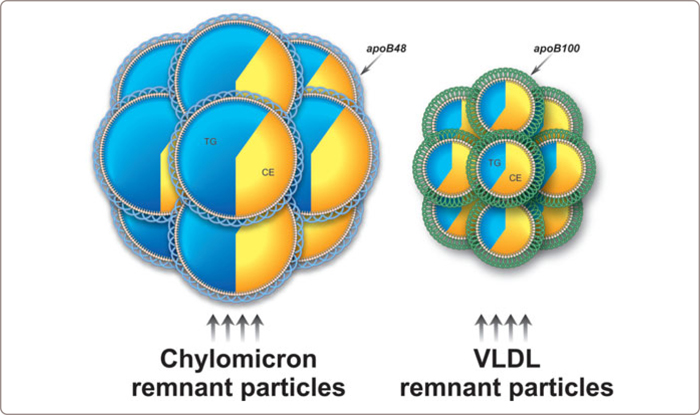
FIGURE 3.12
3.1.3.2 Pathophysiology
As outlined in Chapter 1 , the triglyceride-rich lipoproteins, chylomicrons and VLDL are metabolized in two steps: first, the bulk of the triglycerides are removed in peripheral tissues and second, and almost immediately thereafter, the products of the first step, known as remnant lipoprotein particles, are removed by the liver (See Figure 1.14 , 1.5 and 1.6 ). In Figure 3.13 the normal endogeneous lipoprotein pathway is presented. Because these normal remnant particles are removed so rapidly, they are present in only small numbers in plasma – one-tenth or less the number of VLDL particles in the peak postprandial period. Drastic failure of the second step is the hallmark of this phenotype and leads to the accumulation of modified highly atherogenic remnant particles within plasma – 20 to 40 fold above normal.
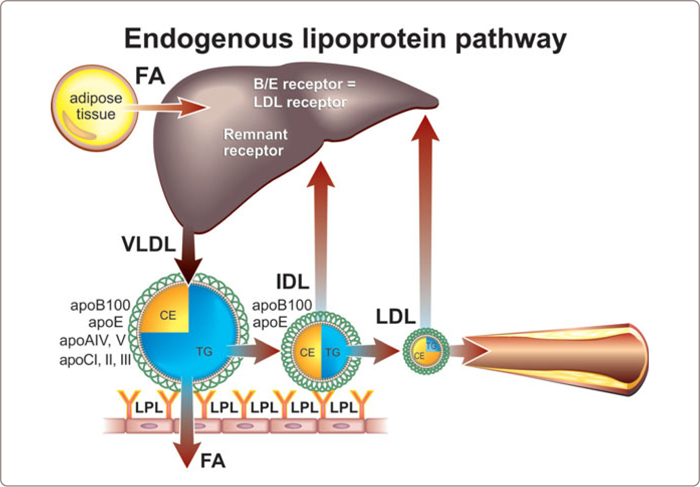
FIGURE 3.13
The normal removal process for chylomicron remnants is complex and has many interconnected parts, including multiple enzymes – hepatic lipase and lipoprotein lipase – multiple proteins with receptor-like functions, such as the LDL-receptor related protein (LRP) and the putative VLDL receptor, and other potentially critical components of the membrane, such as heparin sulfate proteoglycans and apoE. The processes by which VLDL particles are either removed from plasma or converted to LDL particles are even more complex and less well understood. In both instances, it appears that the particles must associate with the external membrane of the hepatocytes and apoE has been thought to be one of the important elements involved in this process.
3.1.3.3 Causes
ApoE2/E2 Mutation: Frederickson, Levy and Lees labelled this disorder type III hyperlipoproteinemia;102 others have designated it familial dysbetalipoproteinemia (FDBL).25,26We prefer the term remnant lipoprotein disorder. Whatever the label, this disorder injures and kills those that it affects and does so rapidly and with no remorse. That is the dark side of this diagnosis. The other side, the bright side, the side of hope, is that this phenotype is eminently treatable, which means that making the diagnosis in time can be life-saving.
Indeed, a characteristic genotype – apoE2/E2– has been thought to be a hallmark of remnant lipoprotein disorder (Figure 3.14 ). ApoE2 has a lower affinity for the hepatic receptor, which removes normal chylomicron and VLDL remnants, than the two other alleles of apoE: apoE3 and apoE4. Other rare mutations in apoE were associated with a dominant form of FDBL where the hyperlipidemia is fully manifest in the heterozygous state. More recently, however, it has become clear that an apoE2/E2 genotype is present only in about 30 % of patients with phenotypic positive remnant lipoprotein disorder indicating the pathophysiology must be reconsidered.25 Actually, it has been known for some time that the apoE2/E2 phenotype alone (approximately 1 % of the population) is not sufficient to produce the full clinical syndrome which is present in only 1/50 or less of E2/E2 individuals. An additional provoking factor must be present to produce the full clinical syndrome. The most common of these are a high-fat diet, diabetes mellitus, obesity, hypothyroidism, renal disease, estrogen deficiency, alcohol use, or certain drugs (Figure 3.14 ).
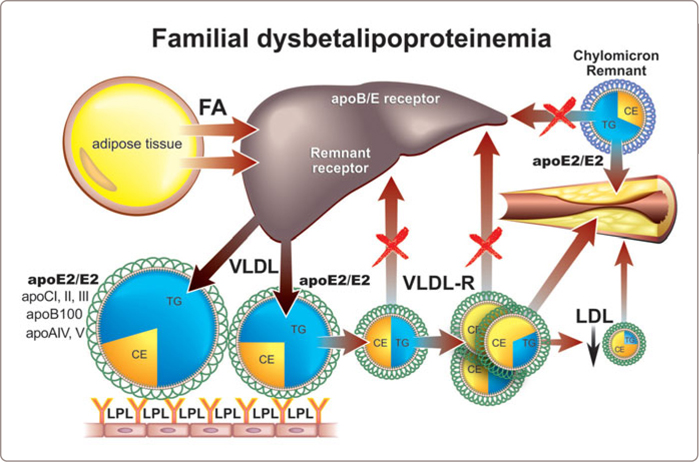
FIGURE 3.14
Whatever the actual reason for their impaired clearance, if VLDL particles are not removed rapidly from the plasma, they necessarily circulate for much longer periods within it and consequently, by virtue of CETP-mediated core lipid exchange with HDL and LDL particles, become cholesterol-enriched abnormal remnant lipoprotein particles (see Figure 1.12 ). Because each abnormal remnant particle contains substantial amounts of cholesterol and triglyceride, marked hypercholesterolemia and hypertriglyceridemia are the lipid hallmarks of this phenotype. Because only a small fraction of VLDL particles is metabolized to produce LDL particles, the number of LDL particles – LDL apoB – is strikingly reduced. The total apoB is characteristically normal due to a reciprocal striking increase in the number of remnant chylomicron and VLDL particles.25,88 Whereas normally, VLDL particles make up only 10 % of total apoB particles and LDL particles make up 90 %, in remnant lipoprotein disorder, LDL makes up approximately 60 % and the remnants 30 to 40 %, so 20-40 fold more than normal. It is the staggering increase in remnant particle number that accounts for the profound increase in cardiovascular risk (see Table 2.3).
3.1.3.4 Clinical Diagnosis
Unfortunately, from the conventional standard panel of lipid tests, the diagnosis can be suspected but not made. These patients have combined hyperlipidemia with classically marked elevations of both triglycerides and cholesterol. Rough equality (if expressed in mg/dl), if present, is suggestive but not much more. However, more recent data demonstrate triglyceride and cholesterol levels may be only moderately elevated.25Moreover, the traditional advanced approaches to diagnosis of this disorder, lipoprotein electrophoresis (broad beta band) or ultracentrifugation (ratio of VLDL-C to total plasma triglyceride > 0.30 mg/dl or > 0.69 mmol/l), are not generally available even in specialty laboratories. ApoE genotyping does identify E2/E2 homozygotes for apoE2. However, absence of the apoE2/2 genotype does not rule out the diagnosis whereas presence of the apoE2/E2 genotype does not mean a particular dyslipidemia is actually remnant disorder. Indeed only a minority of cases of documented remnant disorder are E2/E2 genotype.
Related posts:
 ApoB Lipoprotein Particles: the preferred Treatment Target in Primary and Secondary Prevention
ApoB Lipoprotein Particles: the preferred Treatment Target in Primary and Secondary Prevention
 Secondary ApoB Dyslipoproteinemias
Secondary ApoB Dyslipoproteinemias
 ApoB in Cardiovascular Risk Prediction
ApoB in Cardiovascular Risk Prediction
 The Future of ApoB: Moving from the Risk Prediction Paradigm to the Causal Exposure Paradigm
The Future of ApoB: Moving from the Risk Prediction Paradigm to the Causal Exposure Paradigm
 Diagnosis of the ApoB Dyslipoproteinemias: The ApoB Algorithm
Diagnosis of the ApoB Dyslipoproteinemias: The ApoB Algorithm
 The Life History of ApoB Lipoprotein Particles
The Life History of ApoB Lipoprotein Particles
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


