The inherited disorders of hemoglobin represent the most common monogenic diseases. This article provides a brief description of the main inherited disorders of hemoglobin and their classification, and summarizes progress made in the last decade toward a better awareness and recognition of these disorders as a global health problem. Also presented are the main demographic, genetic, and environmental factors that influence the present and future health burden of these disorders. The strengths and limitations of existing estimates and current health policies in high-, low-, and middle-income countries are discussed.
Key points
- •
The global burden of the inherited disorders of hemoglobin is increasing and this is finally starting to be recognized.
- •
Global, regional, and national newborn and population estimates need to be regularly updated to account for health and demographic changes, and this relies on reliable contemporary epidemiologic data.
- •
The highest burden of the inherited disorders of hemoglobin is and will remain in sub-Saharan Africa for sickle cell disease and in Southeast Asia for the thalassemias, and better prevention and management policies are urgently needed.
Introduction
The inherited disorders of hemoglobin represent the most common monogenic diseases. It has been estimated that around 7% of humans are carrying one of the mutations responsible for these disorders. Although they have been extensively studied at the molecular and cellular level, epidemiologic data to assess the health burden of these disorders are often limited, particularly in regions of high prevalence, which are mostly found in tropical areas. These neglected disorders are becoming an increasing health burden in low-, middle-, and high-income countries. After a short description of the inherited disorders of hemoglobin, this article summarizes progress made toward better awareness and recognition of these disorders as a global health problem before presenting the main factors that influence their present and future burden, and discussing the strengths and limitations of existing estimates.
Introduction
The inherited disorders of hemoglobin represent the most common monogenic diseases. It has been estimated that around 7% of humans are carrying one of the mutations responsible for these disorders. Although they have been extensively studied at the molecular and cellular level, epidemiologic data to assess the health burden of these disorders are often limited, particularly in regions of high prevalence, which are mostly found in tropical areas. These neglected disorders are becoming an increasing health burden in low-, middle-, and high-income countries. After a short description of the inherited disorders of hemoglobin, this article summarizes progress made toward better awareness and recognition of these disorders as a global health problem before presenting the main factors that influence their present and future burden, and discussing the strengths and limitations of existing estimates.
Inherited disorders of hemoglobin
These disorders are classified into two main groups: the structural variants of hemoglobin and the thalassemias, which both follow an autosomal-recessive pattern of inheritance. Maps showing the distribution of each disorder are shown in Fig. 1 .
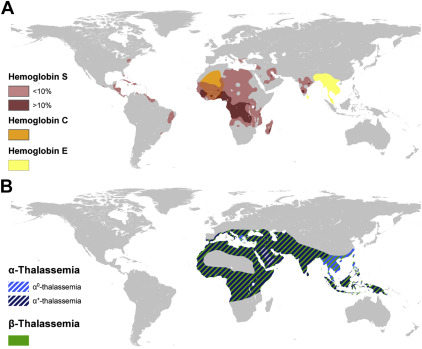
Structural Hemoglobin Variants
More than 1200 structural variants resulting mostly from single amino acid substitutions in the α- or β-globin chains have been identified. Most of them have a limited geographic distribution and reached a low frequency. Nevertheless, the three polymorphisms described next are common and clinically significant.
Hemoglobin S
Hemoglobin S (HbS), also called sickle hemoglobin, is a structural variant of normal adult hemoglobin (HbA) caused by an amino acid substitution at position 6 of the β-globin chain (HBB c.20A > T; p.Glu6-Val). Carriers or heterozygotes (HbAS) are almost always asymptomatic, whereas homozygotes (HbSS) suffer from sickle cell anemia, which often leads to acute and chronic complications including vaso-occlusive crisis, acute chest crisis, or hemolytic crisis. Because of its high level of protection against Plasmodium falciparum malaria, HbS was initially largely restricted to Africa, the Middle East, and parts of India. Nowadays, this variant is found in almost every country, but particularly in the Americas, the Caribbean, and Europe following human diasporas (see Fig. 1 A).
Hemoglobin C
HbC is another structural variant of HbA caused by an amino acid substitution (HBB c.19G > A; p.Glu6Lys) occurring at the same position as HbS. Although carriers (HbAC) are asymptomatic, the inheritance of HbC from both parents (HbCC) causes clinically mild hemolytic anemia. In HbCC, red blood cells have a reduced solubility, which can lead to crystal formation. HbC is mainly of clinical significance when inherited in combination with HbS or with β-thalassemia. HbSC disease causes chronic hemolytic anemia and intermittent sickle cell crises, although slightly less severe or frequent than in HbSS. HbC-β thalassemia leads to moderate hemolytic anemia with splenomegaly. HbC was prevalent only in Western Africa but carriers are now found much more widely (see Fig. 1 A).
Hemoglobin E
HbE is a structural variant of normal hemoglobin (HBB c.79G > A; p.Glu26Lys) affecting the production rate of HbA. Heterozygotes with HbAE are asymptomatic, whereas homozygotes can present some mild clinical features similar to individuals with β-thalassemia trait. Globally, compound individuals with HbE and β-thalassemia represent the highest burden with a wide range of clinical severity. The most severely affected individuals are transfusion-dependent. HbE reaches frequencies up to 60% in parts of Thailand, Laos, and Cambodia, and is highly prevalent in India, Sri Lanka, and Malaysia (see Fig. 1 A).
Thalassemias
The most common forms of thalassemias affect the rate of production of either the α- or β-globin chains that form the subunits of adult hemoglobin, leading to α- and β-thalassemia, respectively. Thalassemias are caused by a large variety of mutations and deletions, causing severity proportional to the inability to synthesize globin chains. Genotypically, both α- and β-thalassemias are classified into minor, intermediate, and major forms. Phenotypically, there is a continuum of severity ranging from asymptomatic to lethal. The geographic distribution of α- and β-thalassemias, which corresponds to the “thalassemia belt” (an area extending from the Mediterranean area through the Middle East and India, to Southeast Asia) is believed to be largely overlapping (see Fig. 1 B). They are now also commonly found in many other parts of the world.
α-Thalassemia
More than one hundred genetic variants of α-thalassemia have been identified. Clinically relevant forms of α-thalassemia usually involve either the deletion of both pairs of α-globin genes on both chromosome resulting in hemoglobin Bart hydrops fetalis syndrome; or the coinheritance of one deleted or inactivated pair on one chromosome (termed α 0 -thalassemia) and a partial production of α-globin chains on the other chromosome (termed α + -thalassemia), caused by deletional or nondeletional variants, and causing HbH disease. Furthermore, mild variants of α-thalassemia act as genetic modifiers of β-thalassemia and HbS. A recent study conducted in Sri Lanka confirmed that substantial variations in the prevalence of α-thalassemia variants can occur over relatively short distances. Because most couples at risk for conceiving fetuses with hemoglobin Bart hydrops fetalis are not currently identified, determining the frequency of the different genetic variants observed in communities and identifying couples who are at risk is urgently needed.
β-Thalassemia
Approximately 250 variants of β-thalassemia identified at the molecular level have so far been shown to reduce the production of β-globin chains. Although most of these are single-nucleotide substitutions, reaching a low frequency within a limited geographic area, a handful of common variants is usually responsible for most of the clinical cases observed in any given region. Patients with the most severe forms of β-thalassemia require regular blood transfusions (typically every 3 weeks) and iron chelation to survive. Stem cell transplantation is the only curative treatment currently available and has been successfully performed in a large number of cases with severe β-thalassemia in high-income countries, but also more recently in some low- and middle-income countries. Screening programs for the prevention and control of β-thalassemia implemented in the 1970s, and often combined with prenatal or premarital diagnosis, have resulted in marked reductions in the birth rate of affected children in Greece, Cyprus, and Italy. Despite high levels of consanguinity, similar results have been reported in several countries in the Middle East following the implementation of large-scale education and counseling programs. However, facilities for simple screening are still lacking in many low- and middle-income countries.
Global health burden
Awareness and Recognition
There is a concerning absence of official recognition of the inherited disorders of hemoglobin as a priority in public health at national, regional, and global scales. Despite being the most common group of monogenic disorders in human populations, few countries have implemented large-scale prevention and management programs for the inherited disorders of hemoglobin and those programs are usually not in place in countries the most affected. This is probably best illustrated by the absence of any national program for sickle cell disease in African countries, whereas universal newborn screening has been implemented in high-income countries, such as the United Kingdom and the United States. We next describe three institutions that have major roles in creating awareness about the inherited disorders of hemoglobin, before listing several initiatives led by the clinical and academic communities to create global and regional networks.
World Health Organization
The World Health Organization (WHO; http://www.who.int/en/ ) adopted two resolutions recognizing the importance of the inherited hemoglobin disorders. In May 2006, the 59th World Health Assembly’s resolution on sickle cell disease (WHA59.20) recognized that “sickle cell anemia is one of the world’s foremost genetic diseases, that it has severe physical, psychological and social consequences for those affected and their families, and that in its homozygote form it is one of the most lethal genetic diseases” and urged Member States to develop and strengthen efforts to prevent and manage sickle cell anemia. This first resolution was further highlighted shortly after at the 118th meeting of the WHO Executive Board by a second one on the thalassemias and other hemoglobinopathies raising concerns about the impact of these disorders on global mortality and morbidity, especially in developing countries, and calling on affected countries and the Secretariat of WHO to strengthen their response to these conditions. In May 2010, the 63rd World Health Assembly adopted an additional resolution on the prevention and management of birth defects, including sickle cell disease and the thalassemias. Through these resolutions, the WHO committed to (1) increase awareness among the international community of the global burden of these disorders, (2) promote equitable access to health services, (3) provide technical support to countries for the prevention and management of these disorders, and (4) promote and support research to improve the quality of life of those affected. In practice, however, the leading role of the WHO to achieve these goals seems to have been quite limited.
Global Burden of Diseases, Injuries, and Risk Factors Study
The Global Burden of Diseases, Injuries, and Risk Factors Study (GBD) is a comprehensive regional and global assessment of mortality (in terms of years of life lost) and disability (in terms of years lived with a disability) from major diseases, injuries, and risk factors (See Kassebaum NJ: The Global Burden of Anemia , in this issue). GBD 2013 produced estimates for 323 diseases and injuries, 67 risk factors, and 1500 sequelae for 188 countries. Disability-adjusted life-years (DALYs) is used as an overall measure of years of life lost and years lived with a disability. The first GBD was published in 1990, which is now used as a reference point for later iterations. GBD 2010 was the first GBD to include the inherited disorders of hemoglobin. Future iterations are expected to be generated on an annual basis. One of the main findings of the GBD is the shift in global health burden from infectious (or communicable) diseases to chronic (or noncommunicable) diseases. GBD 2010 ranked sickle cell disorders 70th (up from 74th in 1990) and the thalassemias 68th (down from 65th in 1990) in terms of DALYs across all ages, respectively. These disorders ranked 17th and 24th, respectively, in the 1- to 4-years age group, reflecting their substantial impact on mortality and morbidity in children younger than 5. GBD 2010 estimated that sickle cell disorders and thalassemias were resulting in 28,640 (confidence interval, 16,756–40,869) and 17,860 (confidence interval, 15,071–20,430) deaths per year, respectively. Although these rankings and estimates are often based on limited epidemiologic data and need refining, they help to show the global health burden associated with the inherited disorders of hemoglobin. Data from GBD 2013 confirm that, whereas the global burden of communicable diseases is decreasing, the inherited disorders of hemoglobin present an increasing burden ( http://ghdx.healthdata.org/global-burden-disease-study-2013-gbd-2013-data-downloads ).
Thalassaemia International Federation
Thalassaemia International Federation (TIF) is a patient-driven organization working in official relations with the WHO since 1996 and with more than 100 national thalassemia associations within 57 countries. Its primary mission is to work toward equal access to quality health care for every patient with a thalassemia syndrome across the world. TIF’s aims to achieve this largely overlap with those of the WHO. In addition to organizing regular global and regional conferences involving clinicians, academics, and patients and patient societies, TIF participates in the formulation and/or amendment of policies in many areas related to the inherited disorders of hemoglobin and actively contributes in adapting these policies to the national level.
Global Sickle Cell Disease Network, Caribbean Association for Researchers in Sickle Cell Disease and Thalassemia, and Sickle Cell Disease Research Network in Central Africa
The Global Sickle Cell Disease Network brings together leading researchers and clinicians in sickle cell disease from high-, middle-, and low-income countries to lead a real change in terms of awareness, research, prevention, and treatment that will not only have a positive impact on children born with sickle cell disease, but also on whole communities and regions. In parallel, several regional networks have been created including, in 2006, the Caribbean Association for Researchers in Sickle Cell Disease and Thalassemia, which brings together 11 countries in the Caribbean region, and in 2010, the Sickle Cell Disease Research Network in Central Africa—Réseau d’Etudes de la Drépanocytose en Afrique Centrale, in which eight Central African countries are involved. Similar networks in West Africa, the Middle East, and India are currently being set up. Such networks aim at stimulating South-South collaborative research projects within regions affected, while promoting awareness and improving prevention and management of the inherited disorders of hemoglobin. Through such networks, policies, methods, and protocols can be standardized, which facilitates epidemiologic studies aiming to assess the burden of these disorders.
Factors Affecting the Health Burden of the Inherited Disorders of Hemoglobin
Malaria selection
The inherited disorders of hemoglobin represent a textbook example of natural selection and balanced polymorphism. It is now well established that they reached high frequencies across large geographic areas because of the protection conferred by heterozygote forms against P falciparum malaria. The improved fitness of those individuals compensates the loss of severely affected homozygotes, allowing the frequency of these disorders to increase. This hypothesis, now commonly called the malaria hypothesis, was first formulated by Haldane in 1949 for the thalassemias and Allison in 1954 for sickle cell disease because of a striking overlap in the geographic distribution of malaria and these disorders. Clear evidence from clinical studies and mechanistic studies conducted in vitro and ex vivo has now added strong support to this hypothesis. Although the mechanisms for malaria protection have been elucidated for some inherited disorders of hemoglobin, controversies remain for others. Recent clinical and theoretic studies have highlighted the importance of epistatic interactions in explaining the frequency and geographic distribution of the inherited disorders of hemoglobin. It is worth keeping in mind that despite considerable efforts to eliminate malaria, this selection mechanism is still ongoing in malarious regions. In nonmalarious regions, whether the mutations have been selected for previously or introduced through population migrations, kinetic models of the spread of genetic disease suggest that declines in the frequency of these disorders are likely to occur very slowly over multiple generations. Studies on sickle cell disease conducted over a couple of decades in Jamaica also showed that substantial improvements in the survival of homozygotes can interfere with the expected decline in allele frequency.
Consanguinity
In many parts of the world, marriages between close biologic kin are preferred for a range of cultural, ethnical, socioeconomic, and religious reasons. Such marriages are categorized as consanguineous when they occur between persons biologically related as second cousins (F ≥0.0156). This is common practice among one-fifth of the world population mostly residing in the Middle East, West Asia, and North Africa, and among emigrants from these communities now residing in North America, Europe, and Australia. From a genetic perspective, consanguinity contributes to increasing the frequency of deleterious recessive mutations. Although consanguinity certainly contributes to increasing the global burden of the inherited disorders of hemoglobin, it is difficult to measure and likely to vary substantially within most countries. Current prevalence data are quite limited and inaccurate, and do not allow to precisely account for the effect of consanguinity on burden estimates.
Population growth
The number of individuals affected by the inherited disorders of hemoglobin is proportional to the overall human population. Based on a total population of 7.3 billion individuals in 2015 and an estimate of 7% carrying one of the mutations underlying these disorders, half a billion individuals either suffer from one inherited disorder of hemoglobin or face the risk of having a child affected. With a world population expected to reach almost 10 billion in 2050 and much faster population growth in tropical and subtropical regions, demography alone provides strong evidence for the increasing burden of this group of disorders. Recent estimates purely based on demographic projections suggested that the annual number of births affected by sickle cell anemia would increase by 33% between 2010 and 2050.
Epidemiologic transition
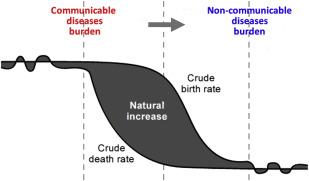
The epidemiologic transition theory assumes that a shift from high birth and death rates to low birth and death rates along with the economic development is also accompanied by a relative increase in the impact of chronic noninfectious diseases in terms of mortality and morbidity ( Fig. 2 ). Through substantial improvements in hygiene, nutrition, public health policies, and infrastructures, partly driven by the Millennium Development Goals, impressive reductions in overall infant and childhood mortality have been observed. As a result, newborns with a severe inherited disorder of hemoglobin who would have previously died undiagnosed are now more likely to survive. As shown in high-income countries, early diagnosis can help prevent serious long-term complications. Nevertheless, such a diagnosis is only meaningful if it is accompanied by access to health care and regular follow-up.
Related posts:
 Global Hematology
Hematological Practice in Hong Kong and China
Glucose-6-Phosphate Dehydrogenase Deficiency
The Present and Future Global Burden of the Inherited Disorders of Hemoglobin
Sickle Cell Disease in Sub-Saharan Africa
Problems and Approaches for Blood Transfusion in the Developing Countries
Global Hematology
Hematological Practice in Hong Kong and China
Glucose-6-Phosphate Dehydrogenase Deficiency
The Present and Future Global Burden of the Inherited Disorders of Hemoglobin
Sickle Cell Disease in Sub-Saharan Africa
Problems and Approaches for Blood Transfusion in the Developing Countries
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


