NORMAL DEVELOPMENT
The placenta is a complex organ that is essential for fetal growth and development. It fulfils a wide spectrum of functions, among which the transport of maternal fuels to the fetus and the synthesis of various hormones and growth factors are the foremost examples. Its development and function are tightly regulated by a range of hormones, cytokines, growth factors, and substrates present in the maternal and fetal circulation. Placenta-derived factors affect the maternal adaptation to pregnancy as well as fetal growth and development.
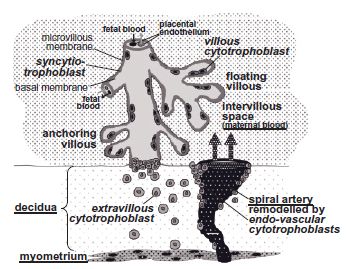
After blastocyst implantation into the decidual surface, the placenta continuously develops by the differentiation and proliferation of trophoblast cells, eventually leading to placental villi of varying degrees of maturation,1 most of which float freely in the intervillous space, i.e. the area between the placental villi (Fig. 3.1). Highly proliferative villous cytotrophoblasts fuse to form the syncytiotrophoblast that represents the outermost interface of the placenta that is in contact with the maternal circulation. The microvillous membrane of this syncytium is in contact with the maternal blood and is richly endowed with receptors,2 enzymes3 and transporters.4 Maternal blood emanating from remodeled and opened spiral arteries bathes the villi.
Some villi physically anchor the placenta to the uterus, thus establishing a connection between the fetus and the maternal decidua (Fig. 3.1). These anchoring villi are formed by proliferation, differentiation, and invasion by cytotrophoblasts of the maternal lining of the decidual cavity. Extravillous cytotrophoblasts also invade the decidual spiral arteries and remodel them into low resistance arteries. The resulting increase of maternal blood flow into the intervillous space ensures adequate maternal nutrient supply to the fetus.1 Trophoblast invasion is tightly regulated in time and space by invasion-promoting and -inhibiting factors originating from the maternal decidua or the placenta. The decidua derives from the maternal endometrium after decidualization and before implantation of the embryo. This process produces a dense extracellular matrix and a cytokine milieu that reduces trophoblast invasion.5 Levels of these factors are altered in various pregnancy-associated pathologies and diabetes mellitus (Table 3.1).
During villous development vasculogenesis and angiogenesis result in the formation of placental vessels, a process that again is controlled by various growth factors, cytokines, and oxygen (Table 3.2), and thus can be dysregulated in diabetes.
Table 3.1 Alterations in maternal levels of trophoblast invasion inhibiting and promoting factors in gestational diabetes mellitus (GDM) and Type 1 diabetes mellitus (T 1 DM). Tumor necrosis factor-α (TNF-α) inhibits trophoblast invasion, whereas vascular endothelial growth factor (VEGF), leptin, insulin-like growth factors 1 and 2 (IGF 1, IGF 2) promote trophoblast invasion.
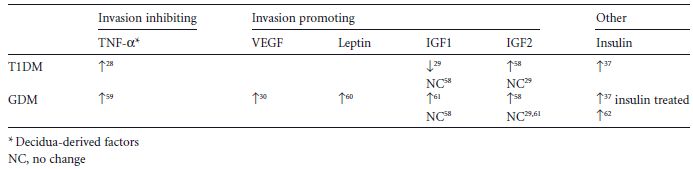
Fig. 3.1 Organization of placental villi after week 20 of gestation. The syncytiotrophoblast represents the outermost surface of the placenta and is in contact with the maternal blood via its microvillous membrane that is richly endowed with receptors, enzymes, and transport molecules. The syncytium regenerates and expands by proliferation and fusion of cytotrophoblasts lying underneath. Some cytotrophoblasts at the tips of floating villi invade the decidua, thereby anchoring the villi. A proportion of these extravillous cytotrophoblasts invade the uterine spiral arteries, leading to their remodeling into low resistance vessels. To differentiate between maternal and placental cells and tissues, maternal structures are dotted and their labelling is underlined.
THE PLACENTA IN DIABETES
Because of the presence of receptors and enzymes on both placental surfaces, i.e. the microvillous syncytiotro phoblast membrane as well as the basal membrane of the syncytiotrophoblast and the placental endothelial cells, the diabetic environment may have profound effects on placental development and function. We recently proposed that these specific effects will critically depend on the time period in gestation when the insult of the diabetic environment acts upon the placenta.6
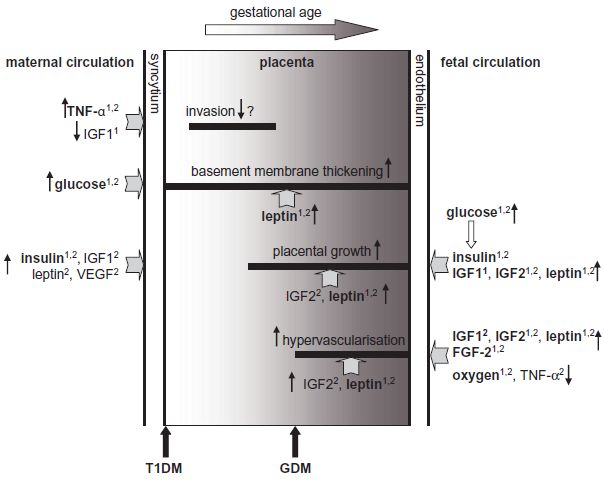
As glucose can stimulate and repress gene expression,7 maternal and fetal hyperglycemia are likely to have an impact on the production of various placental proteins, but a detailed analysis is pending. Moreover, maternal and fetal hyperinsulinemia also affect placental metabolism, growth, and development.3,8,9 However, the changes in the diabetic environment extend beyond glucose and insulin (Tables 3.1 and 3.2). Those in the mother can induce modifications in the placenta, including altered synthesis of cytokines and growth factors, which in turn may act locally in an autocrine or paracrine manner. Altered cytokines and growth factors, along with metabolites, can be secreted into both the maternal and fetal circulation, and thus affect both mother and fetus (Fig. 3.2).
Despite the improvement in maternal glycemic control over the last few decades10 structural and functional changes of the diabetic placenta at term may occur independent of the type of diabetes.11 Similar to fetal weight, placental weight tends to be heavier in diabetes, but the weight gain is more pronounced in the placenta than in the fetus, as is reflected in a higher placental-to-fetal weight ratio than in normal gestation.12,13 It has remained an unresolved question whether placental overweight is the cause or consequence of fetal overgrowth in diabetes.
Intuitively, possible changes in placental transport in diabetes may be implicated. Despite altered expression of placental glucose transporters, perfusion experiments have demonstrated an unchanged if not reduced transplacental glucose transport in GDM14 even on a total placenta weight basis. Studies that also integrate potential structural changes argue strongly for the steeper maternal-to-fetal glucose concentration gradient as the major, if not only, reason for increased glucose fluxes across the placenta in diabetes. This conclusion is also supported by the unchanged concentration differences for glucose in umbilical arteries and veins in GDM.15
Table 3.2 Alterations in fetal levels of pro- or anti-angiogenic factors in pregnancy with gestational diabetes mellitus (GDM) and Type 1 diabetes mellitus (T 1 DM). Both types of diabetes are characterized by enhanced vascularization.
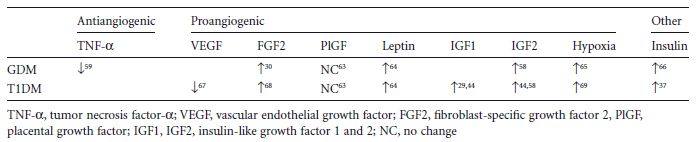
Fig. 3.2 Hypothetical model for diabetes-induced alterations in human placenta. Elevated maternal tumor necrosis factor-α (TNF-α) and reduced insulin-like growth factor 1 (IGF1) levels in Type 1 diabetes mellitus (T1DM) may inhibit placental invasion, paralleling a higher incidence of early pregnancy loss in diabetes. Maternal hyperglycemia induces thickening of the placental basement membrane, hence reducing oxygen transport. Increased levels of placental leptin may even further contribute to the excessive extracellular matrix synthesis. Various factors elevated in the placenta (IGF2, leptin), maternal (insulin, vascular endothelial growth factor [VEGF]) or fetal (insulin, IGF1, IGF2, leptin) circulations in diabetes promote proliferation and placental growth. Placental hypervascularization may be supported by elevated levels of placental IGF2 and leptin, increased fetal IGF1, IGF2, leptin, fibroblast-specific growth factor 2 (FGF2), and reduced fetal TNF-α, as well as by fetal hypoxia. These derangements in the feto-placental compartment are characteristic of gestational diabetes mellitus (GDM), overt diabetes, or both.1 Changed in T1DM;2 changed in GDM. Factors in bold denote similar dysregulation in T1DM and GDM.
Syncytiotrophoblast amino acid transport systems may be altered in diabetes.4,16 However, even for transport systems that are unaltered, when expressed per unit protein or tissue weight, an increase in total placental weight will result in increased nutrient transport. It is unclear if this will stimulate fetal growth or just serve to cover the increased fetal nutrient requirements when the overgrowth of the fetus overgrowth is driven by other factors.
In all types of diabetes, gross placental structure may be altered. In particular, the surface and exchange areas are enlarged17 as a result of hyperproliferation and hypervascularization. The underlying mechanisms for the villous surface increase are not clear. Maternal hyperinsulinemia early in gestation is a candidate,8 but other maternal growth factors may also contribute.
The greater placental capillary surfaces may result from feto-placental counter-regulatory mechanisms to fetal hypoxia which can be inferred from the elevated fetal erythropoietin levels, polycythemia, and increased nucleated red cells often observed in fetuses of women with diabetes.18 Materno-placental oxygen supply may be reduced in diabetes because of:
- Decreased maternal arterial oxygen saturation and increased proportion of glycosylated hemoglobin, which has a higher affinity for oxygen than non-glycosylated hemoglobin19
- Thickening of the trophoblast basement membrane20 although this is not uniformly found21
- Under certain instances, reduced utero-placental blood flow22 as a result of increased flow impedance in the uterine and umbilical arteries.23,24
In addition to impaired oxygen supply, fetal oxygen demand is increased because aerobic metabolism is stimulated by fetal hyperinsulinemia. The resulting low fetal oxygen levels ultimately upregulate the transcriptional synthesis of proangiogenic factors in the feto-placental compartment. Established examples include fibroblast growth factor 2 (FGF2), vascular endothelial growth factor (VEGF), and leptin.25–27 Higher levels of these factors promote placental endothelial cell proliferation a key process in angiogenesis. The increase in placental vascular exchange area against a background of fetal hypoxia appears paradoxical in a situation of maternal nutritional oversupply and may underline the overriding importance of adequate oxygen delivery to the fetus.
Little is known about the placental changes in the first trimester, when the developing placenta is exposed to the maternal diabetic environment, i.e. hyperglycemia, hyperinsulinemia resulting from the relatively excessive insulin doses needed to maintain strict metabolic control, increased expression of TNF-α,28 reduced insulin-like growth factor 1 (IGF1),29 and elevated FGF2.30 It seems reasonable to assume that the diabetic milieu will have an influence on placental development and function during this critical period when placental structures are formed, and the placenta is likely to be most sensitive to environmental derangements. Placental growth and development sometimes appear to be retarded in the first gestational weeks, probably because of a reduction of trophoblast proliferation resulting from hyperglycemia.31,32 A higher incidence of spontaneous abortions33 and pregnancy pathologies, such as pre-eclampsia and intrauterine growth restriction (IUGR), suggest impaired trophoblast invasion, which would result in inadequate placental anchoring and opening of the maternal spiral arteries.34 This is further supported by the reduced utero-placental blood flow, as observed occasionally22 although not uniformly.35,36
Placental expression and activity of the matrix metalloproteinases MMP14 and MMP15, major proteases involved in tissue remodeling processes associated with invasion, angiogenesis, and proliferation, are elevated in Type 1 diabetes3 induced by elevated maternal insulin and TNF-α.28,37 MMP14 and MMP15 accept a remarkably wide spectrum of substrates, including components of the extracellular matrix.38 In addition, mature and immature cytokines may become activated or inactivated, thus further contributing to the alterations in diabetes. In particular, the active form of placental MMP14, which is generated by cleavage by the protease furin, is elevated in diabetes. Furin contains a HIF1-α promoter binding site. This makes it tempting to hypothesize that hypoxic conditions in the villous placental structure in diabetes may be implicated as a cause of increased MMP14 activity. These results demonstrate the sensitivity of early placental development to changes in growth factor and cytokine levels. However, reduced trophoblast invasion in maternal diabetes still remains speculative.
Role of the insulin/insulin-like growth factor system and leptin
Maternal and fetal hyperleptinemia are well-established in diabetes and obesity. Both insulin and leptin fulfil pleiotropic roles beyond the regulation of metabolism, including stimulating growth factor activity and potency, which in turn stimulates expression of various target genes.9,39 Resistance to insulin and leptin occurs often coincidentally in human obesity, because of the considerable overlap between their signaling pathways.40 The extensive cross-talk between their signaling cascades may represent a major contributing factor to the diabetes-induced placental changes, especially in the first trimester of pregnancy.
Insulin, insulin-like growth factor 1 and 2
The insulin/insulin-like growth factor system is thought to have a central role in the control of fetal and placental growth and development.41 The insulin receptor and the highly related IGF1 receptor essentially signal through two main intracellular pathways:42 the extracellular signal-regulated kinase (ERK)1/2 pathway stimulating proliferation and the phosphoinsitide 3-kinase (PI3K)/protein kinase B (AKT) pathway mainly modulating metabolic function.
The feto-placental expression of insulin, IGF1, IGF2, and their receptors is developmentally regulated in a tissue-specific manner and can be affected by nutritional and endocrine conditions.41 Placental expression of insulin receptors undergoes a developmental shift from the trophoblasts in the first trimester to the placental endothelial cells in the third trimester.9,43 The placental IGF1 receptor (IGF1R) is mainly expressed on the basal membrane of the syncytiotrophoblast. Hence, it is predominantly accessible for fetal IGF1 and IGF2.2 The specific roles of these growth factors for the human placenta have not been investigated in great detail. Targeted disruption of the fetal IGF1, IGF2 or IGF1R gene in mice resulted in retardation of fetal growth, whereas IGF2 overexpression enhanced fetal growth. IGF1 stimulates fetal growth dependent on the nutrient supply, whilst placental IGF2 is a key regulator of placental growth and nutrient transfer, thereby allowing enhancement of fetal growth.41
The effects of IGF1 and IGF2 can be attenuated or amplified by soluble IGF-binding proteins (IGFBPs) that influence their bioavailability. In humans, the most prevalent IGFBPs in fetal plasma and tissue are the IGFBPs 1–4. Fetal cord blood data suggest that these binding proteins may be dysregulated in diabetes in pregnancies.44.A decrease in IGFBPs would result in higher bioavailability of IGFs and thus, indirectly might contribute to fetal overgrowth in diabetes.
The endocrine interaction between mother, fetus, and placenta is exemplified by the effect of maternal and fetal insulin on the placenta. Maternal insulin affects placental development3 via receptors expressed on the microvillous membrane of the syncytiotrophoblast. In turn, the placentaaffects the mother by secretion of hormones, cytokines, and metabolic waste products. For instance, maternal insulin upregulates leptin production in trophoblast cells45 and, after secretion into the maternal circulation, leptin enhances maternal insulin resistance. Both leptin and insulin suppress secretion of placental growth hormone (PGH) in trophoblast cells.46 PGH can cause maternal insulin resistance.47 Thus, a reduction of PGH secretion by insulin and leptin may represent a materno-placental forward feedback mechanism, ultimately alleviating maternal insulin resistance.
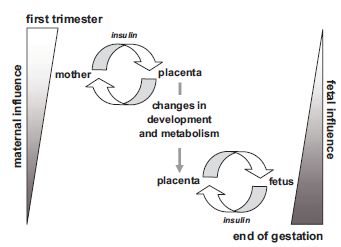
Fetal insulin affects gene expression in endothelial cells from placental arteries and veins,9 which will directly or indirectly affect placental and fetal development. The change of insulin receptor expression from the trophoblast in the first trimester to the endothelium at term thus enables maternal insulin to regulate placental function at the beginning of gestation, whereas as gestation advances, the fetus takes over control of placental insulin effects9 (Fig. 3.3).
IGF1 and IGF2 stimulate trophoblast invasion48 b y upregulation of the metalloproteinases MMP2 and MMP9 that degrade gelatine and collagen, components of the extracellular matrix. Lower maternal IGF1 levels in Type 1 diabetes mellitus may thus contribute to impaired trophoblast invasion. Insulin and IGFs stimulate nutrient transport through the syncytiotrophoblast, in particular the transport of a broad range of neutral amino acids by upregulation of amino acid transporter system A.49–51 Hence, in GDM, transplacental amino acid transport and thereby fetal growth may be promoted by the diabetes-associated increase in maternal concentrations of growth factors (Table 3.1).
Changes can also be seen in the fetal circulation (Table 3.2). However, the consequence of these changes for the fetus, apart from the well-known insulin-stimulated fat accretion, remains unclear.
Leptin
Leptin is a central hormone in metabolic control indirectly promoting insulin resistance.52 In humans, leptin levels correlate highly with adiposity. However, the hormone has various functions beyond metabolic control, such as stimulation of angiogenesis, and regulation of hematopoiesis and the inflammatory response.53 The main source of leptin is adipose tissue, but it is also expressed in various organs of the feto-placental unit. During gestation maternal leptin concentrations rise by 30% and the placenta becomes the primary leptin source.
Fig. 3.3 Spatio-temporal change of insulin receptor expression in the placenta allows a shift in control of insulin regulation from the mother to the fetus. Insulin receptor expression shifts from the trophoblast in the first trimester to the endothelial cells in the third trimester. In the first trimester, maternal insulin influences the placenta by interaction with trophoblast insulin receptors. These may in turn affect the mother by secretion of cytokines, hormones, or metabolic waste products. Later in gestation, the fetus takes over control of insulin-dependent placental processes by fetal insulin interacting with placental endothelial cells. The effects on placental development and metabolism induced at the beginning of pregnancy by maternal insulin along with the effects of fetal insulin on the placenta later in gestation may have repercussions on fetal development and metabolism. (Reproduced from Desoye and Hauguel-de Mouzon,6 with permission of The American Diabetes Association.)