Fig. 2.1
mTOR signaling pathway (detailed in text)
2.2.1.1 mTORC1
mTORC1 includes two unique binding partners: regulatory associated protein of mTOR (Raptor), which recognizes mTOR substrates through their TOR Signaling (TOS) motifs [10–12], and proline-rich protein kinase B (PKB/Akt) substrate 40 kDa (PRAS40), a negative regulator [13, 14]. The most studied effectors downstream of mTORC1 are the 40S ribosomal protein (RP) S6 kinases (S6K1/2), the protein synthesis initiation factor 4E inhibitory proteins (4E-BP1-3), and the autophagy initiating unc-51-like kinases (ULK1/2) (Fig. 2.1). A number of additional mTORC1 substrates have been described in the literature, and their specific roles in cellular processes will be discussed in more detail below (see Sect. 2.4). Additional putative substrates of mTORC1 have been identified in genome wide phosphoproteome screens by quantitative mass spectrometry and require further mechanistic and validation studies [15, 16].
2.2.1.2 mTORC2
mTORC2 is a multiprotein complex in which Raptor is replaced by a large adaptor protein, termed rapamycin-independent companion of mTOR (Rictor). mTORC2 does not signal to either S6K1 or 4E-BP1; is largely resistant to rapamycin, though this view has been recently challenged [17, 18]; and controls actin cytoskeleton dynamics as well as cell survival [19–21]. Other unique binding partners include the mammalian stress-activated map kinase-interacting protein 1 (mSin1) [22, 23] and protein observed with Rictor 1 and Rictor 2 (Protor1/2) [24, 25]. While proline-rich protein 5-like protein (PRR5L) has been reported to bind to mTORC2 through Rictor/mSin1, it is not an essential component of mTORC2 [26]. Presumably, Rictor binds to mTOR at a similar location as Raptor, thereby competing for binding to mTOR [27]. Downstream, mTORC2 regulates the activity of a number of S/T kinases including PKB/Akt [21], glucocorticoid-regulated kinase 1 (SGK-1) [28], and protein kinase C (PKC) [29].
2.2.2 Activation of mTOR Complexes
Both mTORC1/2 complexes respond to hormones and mitogens, but only mTORC1 responds positively to nutrients and energy, including branched chain amino acids (BCAAs) and glucose [30]. In addition, mTORC1 is sensitive to different stresses such as hypoxia and DNA damage.
Most mitogens initiate mTORC1 signaling by the sequential activation of PI3K and PKB/Akt, which reverses the inhibitory effects of Tuberous Sclerosis Complex proteins 1 and 2 (TSC1/2) and PRAS40 on mTORC1 (Fig. 2.1) [13, 31]. TSC1/2, a GTPase-activating protein complex, normally drives the Ras homolog enriched in the brain (Rheb), a small GTPase required for mTORC1 activation, into the inactive GDP state [32, 33], whereas suppression of PRAS40 relieves its inhibitory effect on mTORC1 [13, 34]. It has been reported that Wnt ligands, which regulate cell proliferation, survival, and differentiation [35], positively impinge on mTORC1 through TSC1/2 blockade [36]. Moreover, TSC1/2 appears to act as a node in channeling information from pro-inflammatory signals [37], hypoxia [38–40], or energy stress sensed by AMPK [41, 42]. Importantly, both TSC1/2 and/or AMPK-independent mechanisms of energy sensing and subsequent mTOR inhibition have been established [43, 44]. The Sestrins are another class of metabolic homeostasis regulators which inhibit mTOR signalling at the the TSC1/2 node [45, 46]. Apart from these inputs, DNA damage-induced p53-dependent transcriptional mechanisms downregulate mTORC1/PI3K signaling [47] and also activate AMPK, thus reinforcing negative signaling to mTORC1 [46].
Amino acids and glucose mediate mTORC1 signaling independent of TSC1/2, through the class III PI3K, the human vacuolar protein sorting 34 (hVps34), the Rag GTPases obligate heterodimers (RagA or RagB with RagC or RagD), and a lysosomal docking complex termed Ragulator [30, 48–51]. In the case of the Rag GTPases, in the presence of amino acids, the RagA/B GTPases are GTP charged, which recruits the Raptor-mTORC1 to the lysosomal surface where it can dock at the Ragulator complex and be activated by Rheb [50, 52, 53]. In contrast, RagC/D must be in the GDP-loaded state for mTORC1 to translocate. The hydrolysis of GTP to GDP in RagC/D is achieved through the GAP activity of the folliculin (FLCN) complex and FLCN-interacting protein (FNIP) [54]. mTORC1 lysosomal docking is mediated by either glucose or amino acids and is vital for interaction with Rheb at endomembranes, the location where TSC1/2 signaling also appears to converge with Rheb [55]. Currently, the data support a model whereby the amino acid pool inside the lysosome, and not the cytoplasm, is mediating mTORC1 docking and potential activation. Such sensing appears to be channeled via the lysosomal V-ATPase [56]. ATP hydrolysis by the V-ATPase is necessary for amino acids to promote mTORC1 translocation to the lysosome and subsequent activation [56]. Recently, GATOR 1 and GATOR 2, GTPase-activating complexes, have been shown to drive the RagA/B into the inactive GDP-bound state, thus acting as negative regulators of amino acid sensing [45, 57, 58].
Regulators of mTORC2 have been more elusive. mTORC2 is sensitive to both hormones and growth factors, through a PI3K-mediated signaling pathway [59]. Unexpectedly, it has been reported that the ribosome may also play a crucial role in mTORC2 activation [60]. Ribosomes, but not protein synthesis, are essential for mTORC2 activation, although the mechanism remains unknown. mTORC2 appears to physically interact with ribosomes upon the activation of the PI3K signaling pathway. Conceptually, this may represent a distinct mechanism by which mTOR activation is dependent on favorable growth conditions [60].
mTORC2 was demonstrated to be responsive to insulin, and, in this context, TSC1/2 promoted mTORC2 activation [61, 62], which since surprising as TSC1/2 inhibits mTORC1. Such TSC1/2 regulation of mTORC2 is currently under debate. There are two different models that either advocate for a direct mTORC2 activation by TSC1/2 [61, 63] or an indirect negative feedback loop mechanism that inhibits PI3K signaling, when mTORC1 is further hyperactivated [64] (see Sect. 2.2.3). Nevertheless, some reports also support the existence of PI3K-independent mechanism for activation of mTORC2 [65], including mTORC2’s function in chemotaxis and cytoskeletal organization [66–68]. Recently, Pezze et al. devised a mathematical mTORC1/2 dynamic network model to try and answer which of the several proposed mTORC2/TSC1/2 activation mechanisms, or their interplay, were physiologically relevant [69]. In disagreement with previous models, their data suggest that TSC1/2 is not a direct activator of mTORC2. Although mTORC2 remains PI3K dependent in this model, the signaling to mTORC2 diverges upstream of PKB/Akt [69].
2.2.3 Feedback Loops
The relevance of mTORC1 and mTORC2 as signaling nodes, apart from their nutrient and hormonal inputs, is that both pathways are under control of several negative feedback loops.
2.2.3.1 The S6K1 Negative Feedback Loops
Negative feedback loops are pervasive in biological systems, acting as rheostats which play key roles in cellular homeostasis. These systems ensure that there is no constitutive activation of a given pathway, being responsible for maintaining constant levels of output, as in hormone-mediated protein and lipid production. The inhibitory loops observed in the PI3K/mTOR signaling pathways appear to have evolved to avoid the constitutive activation of anabolic pathways, which if lost may have aberrant consequences at a cellular and/or organismal level [70]. Indeed, studies aimed at inhibiting the mTORC1 and mTORC2 signaling pathways have uncovered several negative feedback loops [70].
It was initially demonstrated through Drosophila genetics that activation of dS6K by dTORC1 unexpectedly dampened dPKB/Akt activation [71, 72]. The activity of the Drosophila orthologue dPKB/Akt is elevated in larvae lacking dS6K or by depletion of dS6K protein levels [71, 72]. Conversely, removal of either dTSC1 or dTSC2, negative effectors of dTOR signaling, led to constitutive dS6K activation and inhibition of dAkt activity. Consistent with these findings, mouse embryonic fibroblasts (MEFs) lacking TSC2 or mammalian cells overexpressing Rheb have constitutive activation of S6K1 and suppression of PKB/Akt activity [32, 73].
S6K1 is not only relevant in protein and lipid synthesis but also responsible for acting upstream of mTORC1/2 signaling at key regulatory points. S6K1 is able to inhibit insulin signaling initiated by the Insulin Receptor Substrate 1 (IRS1). S6K1 promotes multiple site phosphorylation of IRS1 inducing its proteasomal and protein phosphate 2A (PP2A)-dependent degradation, as well as its subcellular relocalization, which feedbacks to suppress PI3K signaling [74–77]. Moreover, these feedback mechanisms do not appear to be limited to the insulin/PI3K signaling, as activation of S6K1 leads to inhibition of the platelet-derived growth factor receptor (PDGFR)-mediated signaling and that of the extracellular signal-regulated kinase/mitogen-activated protein kinases (ERK/MAPK) pathway [78, 79]. PDGFR inhibition impinges on the PI3K/mTOR pathway at the level of PKB/Akt activation, while ERK/MAPK appears to be more complexly and multifunctionally connected to the pathway, including acting through the TSC1/2 node [78, 80–83]. Interestingly, S6K1 has also been implicated in the regulation of mTORC2, by direct phosphorylation of Rictor. However, it is worth noting that this phosphorylation event seems to have few other outcomes than to negatively regulate PKB/Akt phosphorylation at S473 [84].
2.2.3.2 The mTORC2-PKB/Akt Loop
PKB/Akt activation is mainly achieved by PI3K through phosphoinositide-dependent kinase-1 (PDK1) loop phosphorylation of PKB/Akt T308. However, mTOR is also a positive regulator of PKB/Akt through the mTORC2 phosphorylation of PKB/Akt at S473, which in addition to the phosphorylation of T308 is necessary for maximal activation of the kinase [21, 29]. Indeed, mTOR acts functionally downstream and upstream of PKB/Akt. As mentioned above, Pezze et al. [69] recently proposed an mTORC2 activation pathway through a PI3K variant that is insensitive to the negative feedback loop, which regulates mTORC1. This model is contrary to that proposed by Dibble et al. [84]. mTORC2 can also activate SGK proteins, which can mediate PI3K effects independent of PKB/Akt [28, 85].Given that a number of PI3K/mTOR signaling proteins have been reported to be mutated in different tumor types, mTOR inhibitors have been attractive targets in clinical development. Moreover, with the recent epidemiological switch to a more aged society and the onset of the epidemic in obesity, both (i) being mediated by the mTORC1/2 pathways, (ii) having been recognized as key contributors to cancer, and (iii) impinging worldwide, these inhibitors are even more appealing therapeutically [7].
2.3 Inhibitors of PI3K and/or mTOR Signaling
2.3.1 Rapalogs
Rapamycin and its derivatives, everolimus (RAD001) and temsirolimus (CCI-779), termed rapalogs, act by forming an inhibitory complex with the immunophilin FK506-binding protein 12 kDa (FKBP12), which binds upstream of the conserved kinase domain termed the FKBP12-Rapamycin Binding (FRB) domain, thus acting in an allosteric fashion to inhibit mTOR signaling. Although the rapamycins appear to selectively inhibit mTORC1, others have argued that prolonged treatment also leads to inhibition of mTORC2 [86]. Indeed, it has recently been demonstrated that the chronic effects of rapamycin that lead to insulin resistance are mediated by loss of mTORC2 and not inhibition of mTORC1 [17]. The FDA has approved a number of the rapamycins for the treatment of renal cell carcinomas, hormone-receptor-positive/HER2− breast cancers, pancreatic neuroendocrine tumors, and subependymal giant cell astrocytomas. However, the rapamycins can lead to activation of class I PI3K through inhibition of the mTORC1/S6K1 negative feedback loop [71, 87] or to incomplete suppression of mTORC1 signaling to the 4E-BPs [88, 89] and ULK1, both potentially resulting in increased tumor burden. Analyses of patient biopsies treated with RAD001 suggest that activation of PKB/Akt due to loss of the mTORC1/S6K1 negative feedback loop could contribute to tumor progression [90, 91]. Irrespective of treatment response, S6K1-mediated phosphorylation of RPS6 was significantly decreased in matched neuroendocrine tumor and glioblastoma patient biopsies before and after treatment with either RAD001, in combination with octreotide, a somatostatin analog [92], or rapamycin [91], respectively, suggesting effective target inhibition. However, RAD001 affects substrate specificity and not kinase activity. Indeed, RAD001 can abolish S6K1 signaling, while having little impact on other mTORC1 substrates such as 4E-BP1 and ULK1 [18, 93]. Thus, incomplete inhibition of mTORC1 substrates and activation of survival effector PKB/Akt have the potential to lead to drug resistance. Therefore, the new ATP-site-competitive PI3K/mTOR inhibitors should have an added therapeutic advantage by overcoming at least some of the resistance mechanisms induced by rapalogs, as they inhibit the catalytic activity of both mTOR complexes [88, 89, 94] and therefore result in a more complete or durable inhibition of mTORC1/2 signaling.
2.3.2 Dual mTOR and PI3K/mTOR Inhibitors
The new family of PI3K/mTOR inhibitors binds to the ATP-binding pocket of these kinases, inhibiting their activity by competing with ATP [88, 89, 94]. There is an abundance of preclinical data in specific tumor models regarding the impact of dual mTOR and PI3K/mTOR inhibitors both as monotherapies and in combination with other targeted therapies, as well as in combination with chemotherapy and radiation [95]. As single agents, these inhibitors are superior to the rapalogs with regard to inhibition of proliferation and activation of autophagy in vitro and inhibition of tumor progression in vivo (see below). Unfortunately, many of the initial PI3K and/or mTOR inhibitors developed have not survived beyond phase 1/2 clinical trials largely due to no objective tumor responses and/or toxicity mediated by poor formulation, bioavailability, and pharmacokinetics [96–98]. Moreover to date, none have demonstrated superior clinical efficacy over the rapalogs, although theoretically they should revert a number of rapamycin-mediated resistance mechanisms in tumors [90, 91], including PI3K-mediated activation of PKB/Akt and ERK/MAPK signaling [78]. Nevertheless, there are still a number of PI3K and/or mTOR inhibitors being pursued in clinical trials in phase 1/2 for advanced and metastatic cancers either alone or combined with standard and/or targeted therapies (Table 2.1) [99, 100].
Table 2.1
Clinical trials actively recruiting patients for treatment with dual PI3K and/or mTOR inhibitors as single agents or in combination with other therapies (data summarized from clinicaltrials.gov)
Inhibitor | Type of inhibitor | Cancer type | Drugs in study | Clinical trial phase |
|---|---|---|---|---|
AZD2014 | Dual mTOR inhibitor | Prostate cancer (before radical prostatectomy) | Single agent | 1 |
Metastatic or ER+ breast cancers | AZD2014 or everolimus combined with fulvestrant versus fulvestrant | 2 | ||
CC-223 | Dual mTOR inhibitor | Lymphoma, large B cell, diffuse | Combinations of CC-122, CC-223, CC-292, and rituximab | 1 |
Advanced solid tumors non-Hodgkin lymphoma or multiple myeloma | Single agent | 1,2 | ||
GDC-0980 | PI3K/mTOR inhibitor | Advanced or metastatic breast cancer | GDC-0980 or GDC0941 combined with fulvestrant versus fulvestrant alone | 2 |
Castration-resistant prostate cancer (previously on chemotherapy) | GDC-0980 or GDC-0068 combined with abiraterone acetate versus abiraterone acetate | 2 | ||
MLN0128 | Dual mTOR inhibitor | Castration-resistant prostate cancer (previously on chemotherapy) | Single agent | 2 |
GBM or metastatic tumors unresponsive to standard therapy | MLN0128 in combination with bevacizumab | 1 | ||
Advanced non-hematologic malignancies | MLN0128 combined with MLN1117 (oral PI3Kα inhibitor) | 1 | ||
Anaplastic thyroid cancer | Single agent | 2 | ||
PF-05212384 | PI3K/mTOR inhibitor | Advanced cancer | PF-05212384 combined with PD-0325901 (oral MEK inhibitor) or combined with irinotecan | 1 |
Advanced solid tumors | PF-05212384 in combination with either docetaxel, cisplatin, or dacomitinib | 1 | ||
Metastatic colorectal cancer (previously treated 1st line with oxaliplatin-based regimen or have progressed on one) | PF-05212384 plus FOLFIRI. Phase 2 arm will compare PF-05212384 plus FOLFIRI to bevacizumab plus FOLFIRI | 1b,2 | ||
PQR309 | PI3K/mTOR inhibitor | Advanced solid tumors | Single agent | |
SF1126 | PI3K/mTOR inhibitor | Neuroblastoma (pediatric) phase 2 to recruit patients with MYCN amplification or Myc/MycN expression | Single agent | 1,2 |
vs5584 | PI3K/mTOR inhibitors | Advanced non-hematologic malignancies or lymphoma | VS-5584 alone | 1,2 |
Relapsed malignant mesothelioma | VS-5584 combined with VS-6063 (focal adhesion kinase inhibitor) | 1 |
One way to potentially decrease toxicity and/or increase efficacy of PI3K/mTOR inhibitors would be to combine them with a rapalog, since such treatment resulted in a synergistic inhibition of mTOR targets and a significant decrease in tumor progression, in some cases tumor regression, at lower doses of both drugs in a number of mouse models of cancer [18, 101–104]. The increased efficacy at lower doses of both inhibitors is potentially mediated by one drug enhancing the accessibility of the other to its target. Importantly, ATP-site-competitive inhibitors often have off-target effects caused by inhibiting related kinases [105–107]. In contrast, the rapamycins are exquisitely specific in their binding to the FRB domain, immediately upstream of the mTORC1 ATP-binding site [108]. Therefore, this combination should also potentially limit kinase off-target binding.
A second clinical direction being pursued is the use of pan- or isoform-specific PI3K inhibitors [109]. Recently, the PI3Kδ inhibitor has been FDA approved for the treatment of patients with relapsed follicular B-cell non-Hodgkin or small lymphocytic lymphomas [110]. While initial treatment with selective PI3K inhibitors appears to be better tolerated than the pan-PI3K inhibitors, alternative mechanisms of PI3K pathway activation develop and result in dependency on other PI3K isoforms. For instance, some patients treated with BYL719, a specific PI3Kα inhibitor, develop resistance to treatment due to acquired loss of PTEN with corresponding patient-derived tumor xenografts showing response to pan-PI3K or PI3Kβ inhibition [111]. These findings in addition to compensatory induction of a group of receptor tyrosine kinases [112] suggest that neither pan- nor specific PI3K inhibitors would lead to sustained clinical efficacy unless used in combination therapies.
The development of these small molecule inhibitors targeting PI3K/mTOR signaling is not only clinically appealing but has also been crucial in deciphering the mechanisms by which this pathway impacts on different cellular processes leading to human pathologies.
2.4 Impact of PI3K/mTOR Signaling on Cellular Functions
2.4.1 Proliferation
The essential role of mTOR signaling in proliferation and normal development has been clearly demonstrated by various studies using either genetic mutant or knockout (KO) models of key proteins in the pathway and/or treatment with mTOR inhibitors [2, 3, 113, 114]. Mouse KOs for mTOR complex proteins including mTOR, Raptor, Rictor, and mLST8 all die during development at embryonic (E) day ~E5.5, E6.5, E11.5, and E10.5, respectively [4, 5, 17, 115, 116]. The survival of Rictor and mLST8 KO mice to midgestation, longer than either mTOR or Raptor KOs, highlights a differential role of mTORC1 and mTORC2 at different stages of development, and that mLST8 is an essential binding partner of mTORC2 but potentially not mTORC1. Earlier data in Saccharomyces cerevisiae, Drosophila, and mammalian cells revealed that depletion, mutation, or rapalog-mediated inhibition of mTOR and its orthologues resulted in cells accumulating in G1 phase of the cell cycle [3, 114, 117]. Expression of cyclins needed for G1/S transition, including CLN3 or E, was decreased [3, 114]. More recent studies revealed that the effects of mTORC1 on cell proliferation are mediated exclusively by the 4E-BPs [118] and that treatment with PI3K and/or mTOR inhibitors decreased cell cycle progression, eIF4F complex assembly, and abundance of eukaryotic translation initiation factor 4E (eIF4E)-sensitive mRNAs including cyclin D3 [89, 118]. In contrast, the S6Ks were shown to be responsible for the effects on cell size [118], first shown genetically in studies in Drosophila [119]. Also, worth noting is that S6K1 KO mice display a delay in S phase entry following two-third hepatectomy, which can be rescued by in vivo overexpression of cyclin D1 [120].
The characterization of tissue-specific deletions of proteins in mTOR signaling helps delineate the specific role of these proteins in different tissues and is potentially a predictor of adverse effects that may occur when using newer generations of more specific inhibitors to target this pathway. With respect to the proliferative response, KO of Rictor in β-cells decreased proliferation of β-cells and resulted in mild hyperglycemia, while the opposite phenotype occurred in β-specific PTEN KOs [121]. Mice with cardiomyocyte-specific mTOR KO resulted in the death of embryos with mosaic deletion at ~E17.5 due to loss of about half of the cardiomyocytes by apoptosis and decreased compensatory proliferation rates at ~E12.5 [122]. To meet the high demands of proliferation and growth, cells especially tumor cells are dependent on protein synthesis and translation.
2.4.2 Translation
mTOR has been classically described to regulate protein synthesis at translation initiation and more recently at the elongation level, as well as through regulating ribosomal biogenesis [123]. The importance of protein synthesis is evident not only in normal cells during development but also in tumorigenic cells, which require a continuous supply of structural and catalytic proteins. All three steps of mRNA translation are highly regulated, but the majority of the control is argued to be at the rate-limiting initiation step [124–126]. Nevertheless, the ability of a cell to globally increase protein synthesis upon physiological demand is largely accommodated by ribosome biogenesis [123, 127], which, in turn, is highly dependent on RP translation. Indeed, upregulation of enzymatic and structural components of ribosome biogenesis commonly occurs in cells with deregulated proto-oncogenes including Myc, Ras, PI3K, AKT, and mTOR [128–130]. Of note, many of the signaling pathways used by these proto-oncogenes converge on mTORC1. Inhibition of mTORC1, a master regulator of mRNA translation and ribosome biogenesis, has been shown to profoundly change the tumor translational landscape [131]. Intuitively, one might expect that inhibition of mTORC1, a master regulator of protein synthesis, would lead to an increase in total ATP levels as translation is a major energy-consuming process in the cell [132, 133]. However, the contrary was observed in breast cancer cells treated for 12 h with mTOR inhibitors [134]. This is supported by a decrease in the translation of key mitochondria-related mRNAs in an mTORC1-dependent manner and a consequent decrease in mitochondrial activity [134, 135]. mTOR is a key nexus integrating proto-oncogene signaling and nutrient and energy status in order to control the cell’s protein biosynthetic capacity [123].
2.4.2.1 4E-BPs
4E-BPs are known to antagonize the assembly of the multiprotein pre-initiation complex at the mRNA cap by competing with eukaryotic translation initiation factor 4G (eIF4G) for the docking site in eIF4E. The pivotal role of eIF4E is to bind both eIF4G and the mRNA cap to initiate eIF4F complex assembly [125]. mTORC1 negates 4E-BP’s activity by multiple hierarchical phosphorylations that prevent the binding of 4E-BPs to eIF4E [136–139] (Fig. 2.2). However, both the phosphorylation status and the abundance of eIF4E dictate the ability of the 4E-BPs to suppress translation [125]. Noteworthy, the RP transcripts are included in a subset of transcripts controlled by the 4E-BPs, the majority of which belong to the 5′ Terminal Oligopyrimidine tract (5′TOP) mRNA family [131, 140–143]. Although recent studies have suggested a potentially more direct mechanism, with the identification of the La-related family RNA-binding protein 1 (LARP1), which has been reported to be a positive regulator of 5′TOP mRNA stability [144] and required for 5′TOP translational upregulation in an mTORC1-dependent manner [145]. The mitogen-activated protein kinase-interacting kinase 1 and mitogen-activated protein kinase-interacting kinase 2 (MNK1/2) both bind to eIF4G and mediate eIF4E phosphorylation when the initiation factor is recruited to the eIF4F pre-initiation complex [146]. MNK1/2 has been reported to maintain 4E-BP1-independent protein synthesis upon rapalog treatment in the context of rapalog resistance [147]. Thus, through the 4E-BPs, mTORC1 controls global 5′m7G cap-dependent translation and potentially that of the 5′TOP mRNAs.
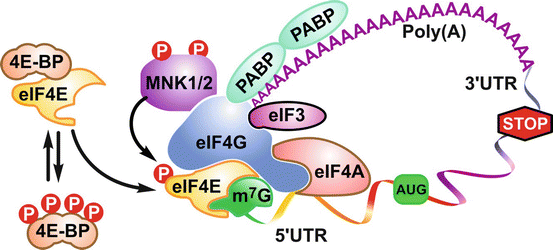
Fig. 2.2
Model for the control of initiation of protein synthesis. In the unphosphorylated state, 4E-BP1 sequesters eIF4E away from the eIF4GI and eIF4GII scaffold proteins, preventing the assembly of a productive eIF4F initiation complex. The critical role played by eIF4GI and eIF4GII is accentuated by their ability to recruit eIF4A, which in turn engages eIF4B, an RNA-binding protein that facilitates eIF4A RNA helicase activity, the poly A binding protein, and MNK1/2 to form a competent eIF4F pre-initiation complex
2.4.2.2 S6Ks
Although S6Ks have less prominent roles in global translation than the 4E-BPs, they appear to regulate protein synthesis by phosphorylating a number of downstream substrates [148]. It has been suggested that, upon activation, mTORC1 is recruited by eIF3 and activates S6K1 that resides in inactive state at the 5’m7G cap of mRNAs. S6K1 would then dissociate from the mRNA and phosphorylate key targets involved in translational initiation, including RPS6, eIF4B, and PDCD4 [149, 150]. It will be of interest to know how lysosomal mTORC1 localization fits in this model. In addition, S6K1 plays a distinct role in the elongation phase of protein synthesis. S6K1 phosphorylates and inhibits the eukaryotic elongation factor-2 kinase (eEF2K), increasing the rate of (eEF2)-mediated translation [151]. Apart from contributing to ribosome production due to its role in translation, mTOR may also impact rRNA synthesis, revealing an even greater control of ribosome biogenesis. Recently, it has been shown that S6K1 can sustain increased pyrimidine biosynthesis and activation of initiation factor 1A (TIF-1A) transcription [152–155], which are indispensable for rRNA transcription. With regard to S6K2, it has been shown to be involved in the 5′m7G cap-independent translation of specific internal ribosome entry site (IRES) containing mRNAs [148], probably through the phosphorylation of IRES-transactivating factors (ITAFs) such as PDCD4 [156].
2.4.3 Metabolism
mTOR actively sustains anabolic metabolism in cells, by driving ATP production both at the level of glycolysis and oxidative phosphorylation. mTORC1 has been implicated in both the transcriptional and translational control of hypoxia-inducible factor 1α (HIF1α), a master regulator of glycolytic genes to increase cellular glycolytic capacity [157–160]. In parallel, the mTOR pathway mediates the upregulation of mitochondrial biogenesis [134, 135]. Although the underlying molecular mechanisms need further elucidation, mTORC1 appears to translationally mediate the expression of a subset of mitochondrial genes through the 4E-BPs. The mTOR-promoted ATP production is mainly used for cell growth and proliferation. Since lipids are the key to membrane biogenesis in proliferating cells, it is not surprising that mTOR is not only a pivotal regulator of protein and RNA synthesis but also of lipid synthesis. Indeed increased lipid production is considered a hallmark of oncogenic proliferation [161].
2.4.3.1 Lipid Metabolism
mTORC1 controls both fatty acid and cholesterol synthesis through its most resonant master genes, the sterol regulatory element-binding proteins 1/2 (SREBP1/2) transcription factors [162]. mTORC1 drives the expression of lipogenic genes by promoting the expression and activation of the SREBPs and by phosphorylating lipin1, a known inhibitor of the SREBPs, thus preventing lipin1 nuclear localization and subsequent protein downregulation of nuclear SREBPs [158, 163–166]. Moreover, mTORC1 promotes adipogenesis through its substrates [167, 168]: (1) S6K1 which regulates the commitment to the adipogenic lineage by regulating the expression of early drivers of adipogenesis [169] and (2) 4E-BPs which exert translational control over PPARγ, a major regulator of adipogenesis [170]. Accordingly, adipose-specific loss of mTORC1 results in lean and high-fat diet (HFD)-induced obesity-resistant mice [171]; S6K1 KO mice are also resistant to age and HFD-induced obesity due to the increase in lipolysis [172] and impairment in adipogenic commitment [169].
A role for mTORC2 in adipogenesis was initially ruled out since adipose-specific Rictor KO mice had no adipogenic impairment [173, 174]. Recent studies support that while mTORC2 may be dispensable in mature adipocytes, it is critical for early adipogenesis through phosphorylation of PKB/Akt on S473 [175]. mTORC2 induces forkhead box protein C2 (FoxC2), a transcriptional factor which inhibits white adipogenesis but promotes brown adipogenesis [175]. Indeed, while mTORC2 loss does not seem indispensable for muscle development and regeneration, it is essential for brown adipose tissue growth, which also arises from the Myf5 mesenchymal lineage, unlike white adipose tissue [176]. In the muscle, mTORC1 is responsible for stimulating protein synthesis necessary for muscle hypertrophy in response to contraction [177]. Loss of muscle mTORC1, but not mTORC2, leads to reduced muscle mass and oxidative function which is eventually lethal [178].
2.4.3.2 Energy Balance Regulation
Apart from directing the whole organism to store excess energy, mTOR also mediates food intake. mTORC1 exerts whole-body energy balance regulation, in the hypothalamus, where it reduces food intake through mechanisms that act to inhibit S6K1 [179–181]. Moreover, in HFD conditions, leptin is unable to activate hypothalamic mTORC1 and/or reduce food intake. This suggests that mTOR deregulation may be implicated in hypothalamic leptin resistance, i.e., deregulated food intake and/or appetite control [179, 180]. At the level of the liver functions, mTORC1 restricts ketogenesis necessary to support peripheral tissues during states of fasting. Therefore, mTORC1 activity is low during fasting, and the inhibition of mTORC1 is required for the fasting-induced activation of peroxisome proliferator-activated receptor α (PPARα), the master transcriptional activator of ketogenic genes [182]. mTORC1 regulates PPARα expression and activity by an S6K2-dependent mechanism which promotes nuclear localization of the nuclear receptor corepressor 1 (NCoR1) [183].
2.4.3.3 Metabolic Syndrome
mTORC1 is highly active in tissues of obese and high-fat-fed rodents [172, 184, 185], which may be a hallmark in the metabolic syndrome. At first, activation of mTORC1 leads to an increase in β-cell size and number, which translates into systemic hyperinsulinemia and glucose tolerance [186, 187]. These effects in part are mediated by S6K1 [188]. However, sustained mTORC1 activation promotes insulin resistance in the adipose tissue, muscle, liver, and β-cells through S6K1-mediated silencing of insulin receptor signaling [7]. mTORC1 is also a positive regulator of pancreatic endocrine function. Impaired insulin signaling in the liver further contributes to the syndrome by upregulating gluconeogenesis, while in the pancreas, it drives the progression into insulin resistance states or diabetes type 2 by promoting β-cell loss [187, 189]. The liver is particularly vulnerable to ectopic fat accumulation, and fatty liver leads to metabolic syndrome. In such a scenario, mTOR activation has been suggested to drive liver lipogenesis through activation of the SREBPs [166, 190]. Consistent with these findings, liver-specific depletion of S6K1 has been shown to protect against HFD-induced hepatic steatosis and systemic whole-body insulin resistance, the latter being associated with reduced insulin levels and loss of the negative feedback loop in the muscle and fat [191].
Noteworthy, rapamycin as a therapy for metabolic syndrome has failed [192]. Some studies have pointed out that rapamycin treatment leads to impaired glucose homeostasis due to (i) β-cell toxicity [193] and (ii) incomplete insulin-dependent inhibition of hepatic gluconeogenesis, which may, controversially, be due to mTORC2 degradation [17].
2.4.4 Autophagy
mTOR regulates autophagy by phosphorylating both positive and negative regulators of this response. The cross talk between ULK1s as a substrate of both mTOR and AMPK is the most investigated to date with respect to autophagy. In nutrient-replete conditions, mTOR inhibits autophagy primarily through phosphorylation of ULK1 on S758 (S757 residue in mice) [194, 195], activating molecule in Beclin1-regulated autophagy 1 (AMBRA1) [196] and Atg13, the latter which is found in a multiprotein complex essential for autophagosome formation and includes ULK1, FAK family-interacting protein of 200 kD (FIP200) and autophagy-related protein 101 (Atg101) [49–51]. mTORC1 has also been reported to phosphorylate ULK1 on S637 [194], a site shared with AMPK [197], which potentially impacts the speed at which cells sense nutrient availability. Not only does mTORC1 regulate the activity of ULK1 by direct phosphorylation, it also impacts its ubiquitylation and stability by phosphorylating AMBRA1 on S52 [196].
It is also known that mTOR controls autophagy at a transcriptional level by regulating the cellular localization of transcription factor EB (TFEB) through phosphorylation of S211. Phosphorylated TFEB is sequestered in the cytoplasm in complex with 14-3-3 (YWHA) proteins thus preventing its nuclear localization and transcription of TFEB target genes involved in autophagy and lysosomal function [198]. The failure to inhibit mTOR signaling and activate autophagy is detrimental, as knock-in mice with constitutive expression of RagAGTP die within their first day after birth due to activated mTOR signaling and therefore failure to activate autophagy for de novo glucose synthesis [51]. Indeed this phenotype of the RagAGTP knock-in mice and postnatal death within 1 day is similar to that of knockout mice for autophagy genes including Atg5 [199], Atg7 [200], and Atg3 [201].
Conversely, inhibition of mTOR either pharmacologically or by “nutrient deprivation” leads to induction of autophagy. TSC2 and Raptor, both downstream targets of AMPK, are key components of the mTORC1 pathway that are critical for AMPK-mediated inhibition of mTORC1 and cell growth in conditions of low energy [41, 44] (Fig. 2.1). When activated, AMPK directs the reprogramming of catabolic processes to maintain ATP levels, while turning off anabolic processes, including carbohydrate, lipid, protein, and rRNA biosynthesis [202]. Despite their critical role in regulating energy, it is worth noting that in MEFs lacking both catalytic subunits (α1 and α2) of AMPK [203], there is no significant difference in ATP levels as a response to energy stress induced by biguanide treatment [43]. Under glucose deprivation, activation of AMPK leads to phosphorylation of ULK1 on S317 and S777 and activation of autophagy [195]. It has been postulated that mTOR phosphorylates the death-associated protein 1 (DAP1), a negative regulator of autophagy, to prevent excessive activation of autophagy under starvation conditions [204].
In a tumor setting, the role of autophagy activation as a result of targeting the PI3K/mTOR signaling is complex, with it either acting as a tumor suppressor or a survival mechanism depending on the tumor type, the stage of the disease, and the cell populations within each tumor [18, 205–207]. For instance, although the combination of a rapalog with a dual PI3K/mTOR inhibitor caused tumor regression in a mouse models of spontaneous hepatocellular carcinoma, presumably through increased autophagy [18], it has been argued that a small population of stemlike cells are protected by mTOR inhibitors and persist, with their survival dependent on autophagy [206, 208], while others have reported that mTOR inhibitors suppress cancer stem cell proliferation, survival, and clonogenic sphere-forming ability of tumors developed in the colon [209], prostate [210], small-cell lung cancer [211], and glioblastoma [212].
2.4.5 Differentiation
Although significant strides have been made in our understanding of the role of mTOR signaling in protein translation, cell growth, and proliferation in adult/differentiated cells [59, 213], little information is available concerning these responses in cancer stem cells (CSCs). In embryonic stem cells (ESCs), global translation rates were found to be reduced by translational regulators, including mTOR and 4EBP1, as compared to differentiated cells obtained from mouse ESC-derived embryoid bodies [214]. Consistent with this finding, it is known that suppression of global protein synthesis rates is essential to maintain ESC in the pluripotent state [215, 216]. Moreover, hyperactivation of S6K1 drives pluripotent stem cells to differentiate [217], and persistent mTOR signaling leads to a reduction in the adult stem cell population of the epithelial compartment of the skin [218]. More recently, inhibition of mTORC1 led to growth arrest and differentiation of established mouse intestinal adenomas by a mechanism involving eEF2K and control of translational elongation [219]. Seemingly consistent with these findings, an inverse relationship has been reported between DEPTOR, a negative regulator of both mTORC1 and mTORC2, and differentiation of ESCs [220]. Together, these results indicate that the control of mTORC1 signaling is critical for the maintenance of pluripotency in ESC.
The role of mTOR signaling in CSCs is controversial. On one hand, persistent mTOR signaling has been shown to maintain the self-renewal and tumorigenicity of glioblastoma stemlike cells and breast cancer stem cells [221, 222]. On the other hand, inhibition of mTOR signaling by rapamycin is leading to the upregulation of cells expressing CD133+, a cell surface marker for CSCs, in tumors including HCCs [223]. This pro-tumorigenic role of rapamycin is supported by recent studies demonstrating that inhibition of mTORC1 significantly enhanced the generation and maintenance of CD133+ CSC population and promoted secondary tumor propagation of H-Ras-transformed mouse HCC cells in vivo [224].
Genetic studies in C. elegans [1], Drosophila [2], as well as in mice [4] have demonstrated that the TOR orthologues, cTOR, dTOR, and mTOR, respectively, play an essential role in the development, which is tightly linked to nutritional status. Thus, to orchestrate the control of homeostatic responses, mTORC1 integrates signals from growth factors and hormones, including insulin, with those emanating from nutrients, including glucose, amino acid, and fatty acids [59, 213]. The insulin and TOR pathways have been implicated in the effect of diet on stem cell proliferation in several contexts, including Drosophila germ stem cells [225, 226], intestinal stem cells [227, 228], and neural stem cells [229, 230]. Accordingly, although some aspects of the cellular and molecular mechanisms linking diet to stem cells may be context specific, their dependence on the nutrient-responsive “insulin-like” and TOR signaling pathways appears to be conserved. For instance, S6K1-deficient mice show reduced ability to accumulate fat and, when challenged with a high-fat diet, demonstrate a striking reduction in the number of early adipocyte progenitors [169]. Also, although S6K1 is dispensable for terminal adipocyte differentiation, it is required for the commitment of ESC to the early adipocyte progenitor lineage and plays a dominant role over the 4E-BPs in adipogenesis [169]. Deletion of Raptor in mesenchymal stem cells decreased differentiation into adipocytes and promoted osteogenesis [231]. It is important to note that mTORC2 had an opposing role with regard to the fate of the differentiation of these stem cells [231]. This opposing role of mTORC1 versus mTORC2 on differentiation of stem cells has also been reported for oligodendrocyte differentiation in the central nervous system [232].
2.5 Summary
The role of mTOR signaling is essential throughout life from development to aging. The functional deregulation of this signaling pathway leads to diseases including diabetes and cancer. Fortunately, inhibitors of this pathway exist and important strides have been achieved to test them clinically with some currently being FDA approved. However, improved therapies leading to sustained clinical efficacy have yet to be attained; potentially using combination therapies targeting a specific patient population, based on driver mutations in specific cancer types, would be more appropriate. The increasing mechanistic understanding of the PI3K/mTOR signaling pathway and its feedforward and feedback processes should lead to improving the design of therapeutic strategies to target disease states while accounting for compensatory mechanisms arising from the pathway’s downregulation.
Acknowledgments
We are thankful to G. Doerman for the assistance with preparation of figures. H.E.T is supported by a faculty pilot project grant by the Department of Internal Medicine, University of Cincinnati, OH. G.T. is supported by grants from the Instituto de Salud Carlos III (IIS10/00015), the Spanish Ministry of Science and Innovation (SAF2014-52162), the CIG European Commission (PCIG10-GA-2011-304160), and the NIH/NCI National Cancer Institute (R01-CA158768). S.C.K is supported by grants from the Instituto de Salud Carlos III (IIS12/00002) GT and SCK are also supported by the European Regional Development Fund FEDER in Spain and from the Spanish Ministry of Economy and Competitivity (BFU2012-38867).
References
1.
Long X, et al. TOR deficiency in C. elegans causes developmental arrest and intestinal atrophy by inhibition of mRNA translation. Curr Biol. 2002;12(17):1448–61.PubMed
2.
Oldham S, et al. Genetic and biochemical characterization of dTOR, the Drosophila homolog of the target of rapamycin. Genes Dev. 2000;14(21):2689–94.PubMedCentralPubMed
3.
Zhang H, et al. Regulation of cellular growth by the Drosophila target of rapamycin dTOR. Genes Dev. 2000;14(21):2712–24.PubMedCentralPubMed
4.
Gangloff YG, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24(21):9508–16.PubMedCentralPubMed
Related posts:
 The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
The Role of mTOR Inhibitors in Neuroendocrine Tumors
The Evolving Role of Mammalian Target of Rapamycin (mTOR) Inhibitors in Renal Cell Carcinoma
The Role of PI3K/AKT/mTOR Inhibitors in the Treatment of Hematological Malignancies
Potential Future Indication of Rapamycin Analogs for the Treatment of Solid Tumors
 Predictive Biomarkers of Response to mTOR Inhibitors
Predictive Biomarkers of Response to mTOR Inhibitors
 Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Rational Combinations of mTOR Inhibitors as Anticancer Strategies
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


