The Perception of Pain
This section is designed to provide a background for the understanding of the anatomy, physiology, and psychology of pain. According to the classical Cartesian model, pain is a hard-wired sensory system in which noxious input is passively transmitted along sensory pathways to the brain. This is an inadequate model for nociceptive information processing. Pain perception is subject to substantial pro- and antinociceptive modulation at multiple levels in the central nervous system (CNS). Pain is a conscious experience, an interpretation of the nociceptive input that is influenced by memories, emotions, pathology, and genetic and cognitive factors.1 Pain is not necessarily related directly to nociceptive drive or input; neither is it solely for vital protective functions. Contrary to previously held beliefs that pain resulted from a central summation in response to excessive sensory stimulation, it is now clear that many factors influencing pain perception are centrally mediated.1 The astute clinician must ascertain the balance between peripheral and central influences and identify those that are caused by pathological as opposed to emotional or cognitive influences. Treatments must be targeted at the relevant causative factors.
NOCICEPTIVE TRANSMISSION
Nociceptive transmission represents the synaptic transfer of information about the intensity, duration, and location of peripheral noxious stimuli from nociceptive afferent axons to dorsal horn neurons. Nociceptors detect noxious stimuli and initiate the peripheral pathway to nociceptive pain. The nociceptor is highly modifiable in response to injury of its axon and to exposure to inflammation.2 Some nociceptors are thinly myelinated (Aδ-fibers) but most are unmyelinated (C fibers), and these slowly conducting afferents represent the majority of sensory neurons in the peripheral nervous system. The nociceptor has four major functional components: the peripheral terminal that transduces external stimuli and initiates action potentials, the axon that conducts action potentials, the cell body that controls the identity and integrity of the neuron and is its metabolic factory, and the central terminal that forms the presynaptic element of the first synapse in the sensory pathway in the CNS (Fig. 3.1). Nociceptive neuron sensitivity is modulated by a large variety of mediators in the extracellular space. These mediators activate a large number of receptor classes, which in turn activate a plethora of signaling cascades (Fig. 3.2).3 A variety of ion channels implicated in nociception are expressed in sensory neurons (Fig. 3.3). In disease states, various ion channels are activated by inflammatory signals and these signals sensitize sensory neuron channels, causing primary hyperalgesia. K+ channels are main ion channels that stabilize the membrane potential by hyperpolarization. The role of the K+ receptor in nociception is not well understood, but it is implicated because of its modulating potential.
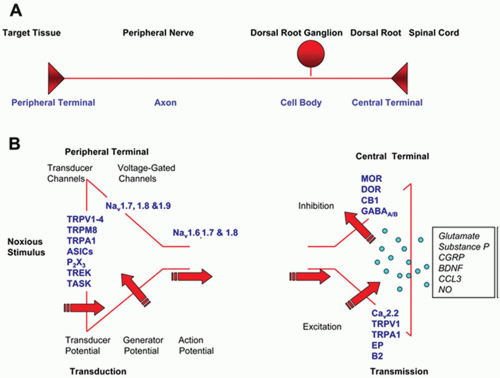 FIGURE 3.1 The nociceptor. A: Operational components of the nociceptor. B: Transduction is mediated by high-threshold transducer ion channels, which depolarize the peripheral terminal activating voltage-dependent sodium channels. Transmission occurs in response to calcium influx at the central terminal, releasing glutamate as well as multiple synaptic modulators and signaling molecules, and is subject to both excitatory and inhibitory influences. (From Woolf CJ, Ma Q. Nociceptors—noxious stimulus detectors. Neuron. 2007;55(3): 353-364. Copyright 2007, with permission from Elsevier.) |
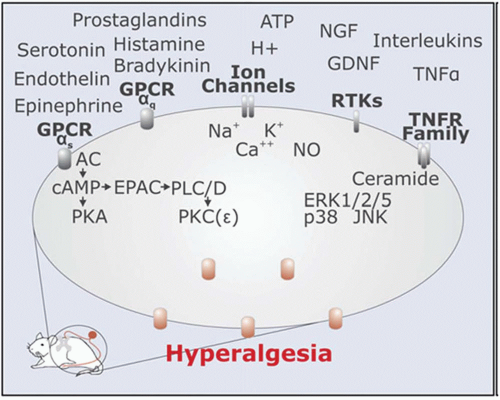 FIGURE 3.2 Signaling components in nociceptors. (From Hucho T, Levine JD. Signaling pathways in sensitization: toward a nociceptor cell biology. Neuron. 2007; 55(3):366. Copyright 2007, with permission from Elsevier.) |
Voltage gated ion channels include Na+, and Ca2+ channels. Na+ channels are classified as tetrodotoxinsensitive or -resistant. There are ten Na+ channel genes, and these channels are responsible for rapid depolarization. Some voltage-gated Na+ (Nav) channels are involved in neuropathic pain.4 Experiments with transgenic mice lines have clearly implicated Nav1.7, Nav1.8, and Nav1.9 in inflammatory pain, and possibly in neuropathic pain.5
There are several different types of voltage-dependent Ca2+ channels (L, N, P/Q, and R). Voltage-dependent calcium channels are formed as a complex of several different subunits (α1, α2δ, β1-4, and γ). Gabapentin is known to interact with the α2δ subunit.6 Nociceptive neurotransmitters such as substance-P and CGRP are released upon activation of L-, N-, and P/Q-type Ca2+ channels.
Transient receptor potential (TRP) ion channels are nonselective cation channels with variable permeability to Ca2+; they are grouped into six families.7 These families are classical (TRPC), vanilloid receptor-related (TRPV), melastatin-related (TRPM), mucolipins (TRPML), polycystins (TRPP), and ankyrin transmembrane protein-related (TRPA). TRP channels play important roles in sensation, because they detect a variety of stimuli, including vision, taste, smell, hearing, mechanosensation, thermosensation, and pain. For example, TRPV1 is activated by capsaicin, noxious heat (>43°C), and acid (a frequent stimulus in inflamed and ischemic tissues). Clones of TRPV1, 2, 3, 4 respond to noxious stimuli. The acid-sensing ion channel (ASIC) is activated by extracellular acid, like TRPV1. Currently, five ASIC genes are known in mammals (ASIC1-ASIC5). ASIC subtypes are modulated by NSAIDs.8 Purinergic receptor subtype X channels (P2X) are adenosine triphosphate (ATP)-gated and have seven subtypes. P2X levels increase with inflammation9 and may play a role in neuropathic pain.10 Serotonin (5-HT) has several G protein-coupled metabotropic receptor subtypes and one ionotropic receptor (5-HT3R). 5-HT3R is involved in activation of nociceptor neurons by serotonin.11
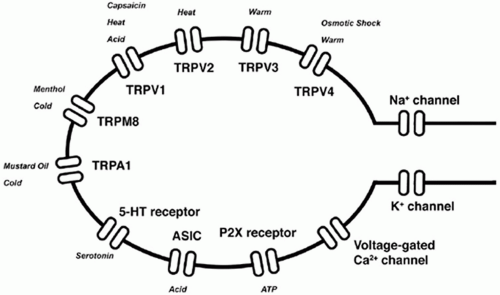 FIGURE 3.3 Ion channels expressed in sensory neurons. (From Lee Y, Lee CH, Oh U. Painful channels in sensory neurons. Mol Cells. 2005;20:315-324.) |
Peripheral sensitization involves a lowering of the threshold of the nociceptor in response to inflammatory sensitizers. These activate the trafficking of sodium channels via diverse signal transduction pathways in the peripheral terminal, largely as a result of phosphorylation.2 Phenotypic switches occur in nociceptors in response to inflammation and axonal injury, by virtue of exposure to retrograde transported signal molecules or the absence of target derived signals. The nociceptor neuron is highly specialized to respond only to noxious stimuli and to communicate this information accurately to the CNS. It is, however, not a static detector; the functional and chemical plasticity of the receptor ensures that its threshold and responsiveness, as well as the efficacy of its synaptic contacts, are regulated to reflect changes produced by activity, inflammation, and axonal injury. Heightened pain sensitivity can contribute to healing by helping avoid contact with the damaged body part until repair has occurred. Persistent alterations in nociceptors that drive pain in the absence of noxious stimuli or inflamed or damaged tissue represent, by contrast, a pathological change in the nociceptive system. Successful treatment of such pain requires restoring the nociceptor to its normal function.
With repeated peripheral noxious stimulation, the trigger for the change in receptive field properties is recruitment of receptive fields outside of this region, resulting in changes within the CNS and not an increased sensitivity of the peripheral terminals of sensory fibers innervating injured tissue.2 Increased excitability triggered within the spinal cord by peripheral noxious inputs represents central sensitization, a state in which the response to normal inputs is greatly enhanced (Fig. 3.4). As such, pain does not simply reflect the presence, intensity, or duration of specific nociceptive stimuli in the periphery, but also changes in the function of the CNS.
Related posts:

Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


