The pathophysiologic consequences of transfusional iron overload (TIO) as well as the benefits of iron chelation therapy are best described in thalassemia major, although TIO is increasingly seen in other clinical settings. These consequences broadly reflect the levels and distribution of excess storage iron in the heart, endocrine tissues, and liver. TIO also increases the risk of infection, due to increased availability of labile iron to microorganisms. The authors suggest that extrahepatic iron distribution, and hence toxicity, is influenced by balance between generation of nontransferrin-bound iron from red cell catabolism and the utilization of transferrin iron by the erythron.
Key points
- •
The pathophysiologic consequences of transfusional iron overload (TIO) are best understood in thalassemia major (TM) and broadly reflect the distribution of excess storage iron to heart, endocrine tissues, and liver.
- •
The pattern of excess iron distribution reflects the pattern of nontransferrin-bound iron (NTBI) uptake to these tissues.
- •
Storage iron does not directly damage cells but its intracellular turnover contributes to labile intracellular iron pools that generate harmful free radicals.
- •
TIO also increases the risk of infection due to increased availability of labile iron to microorganisms.
- •
In other conditions such as sickle cell disease, Diamond-Blackfan anemia, and myelodysplastic syndrome, the propensity to the extrahepatic iron distribution and its consequences vary compared with TM.
- •
The mechanisms underlining this variability may reflect differences in the transfusional iron loading rates, age of commencing transfusion, as well as differences between transferrin iron utilization and NTBI generation.
Iron homeostatic mechanisms
Iron homeostatic mechanisms are key to the pathophysiology of transfusional iron overload (TIO). In humans, these mechanisms are best adapted to increasing iron acquisition in conditions of iron deficiency or anemia, or to limiting iron distribution from the macrophage system during inflammation. They are not well adapted, however, to controlling the distribution of TIO or to eliminating excess iron. This is in marked contrast to rodents, where most studies on iron overload and iron metabolism have been performed and where iron overload is eliminated efficiently by the biliary route. Iron homeostasis is adapted to supplying only that which is essential for the functioning of proteins involved in oxygen transport, oxidative energy production, mitochondrial respiration, and DNA synthesis, while minimizing the potential for iron toxicity from its redox cycling. These homeostatic mechanisms work at 2 levels: firstly at the level of the whole body through interactions of plasma hepcidin with membrane ferroportin and secondly at a cellular level through interaction of iron responsive element (IRE)-binding proteins (IRPs) with IREs present on mRNAs of key iron metabolism–related proteins.
Body Iron Homeostasis
A healthy human contains 40 to 50 mg/kg of iron, mainly as hemoglobin (30 mg/kg). About 4 mg/kg is present in muscle myoglobin, with 2 mg/kg in cells as iron-containing enzymes. Storage iron, present as ferritin and its compact, partially degraded form hemosiderin, ranges from 0 to 2000 mg ; this is mainly present in liver, spleen, and bone marrow (BM) macrophages, formerly referred to as the reticuloendothelial system (RES), and in hepatocytes. Liver iron concentration (LIC) rarely exceeds 1.8 mg/g dry weight (dw) in healthy individuals in the absence of liver disease, hemochromatosis genes, inappropriate dietary supplementation, or blood transfusion.
A healthy individual absorbs only about 10% of dietary iron or about 1 to 2 mg/d, usually balanced by iron loss from skin, gut, menstruation, or pregnancy. Anemia, hypoxia, ineffective erythropoiesis (IE), and the presence of variant HFE genes increase iron absorption, the common factor being low, or inappropriately low, plasma hepcidin levels. The latter permits higher enterocyte ferroportin expression, allowing Fe(II) flux and hence increasing dietary iron absorption. Iron absorption is also increased through hypoxia-inducible factor 1–mediated signaling, by duodenal upregulation of DcytB and DMT1 expression. Thus, in principle, any anemia will tend to increase the efficiency of iron absorption. Most body iron turnover, however, is not directed through iron absorption, but through plasma transferrin, which, although binding only 1 to 2 mg of iron at any moment, in a healthy adult delivers about 20 to 30 mg/d via transferrin receptors on the erythron for hemoglobin synthesis.
Hepcidin regulation is important both to iron absorption from diet and to iron egress from erythrophagocytic macrophages. Hepcidin controls iron egress from both macrophages and enterocytes by binding to and degrading ferroportin, through which Fe(II) exits these cells. Hepatic hepcidin synthesis is controlled by at least 3 distinct regulatory mechanisms responsive to levels of iron, erythropoiesis, or inflammation.
- •
Extracellular iron sensing involves the binding of diferric transferrin to transferrin receptor 1 (TfR1), initiating the translocation of HFE from TfR1 to TfR2 and its subsequent signaling via ERK1/ERK2 and p38 MAP kinase to induce hepcidin expression. Storage iron sensing is affected by BMP6 signaling via BMP receptor (and SMADs pathway) whose sensitivity is markedly increased by its interaction with hemojuvelin, HFE, and TfR2 in holotransferrin-dependent manner, thus enhancing hepcidin transcription.
- •
Erythropoiesis sensing involves BM-derived factors that suppress hepcidin synthesis; conditions with high levels of IE will have high levels of these factors, the nature of which has been debated. These include GDF15 ; twisted gastrulation factor-1 ; and most recently erythoferrone, which has been identified as a key factor in mice, although its relevance in humans has yet to be demonstrated. Another separate erythropoiesis sensing mechanism likely involves desaturation of transferrin by the erythron as disruption of transferrin iron uptake into erythron in hbd mouse increases hepcidin level despite ongoing anemia, presumably overriding the erythropoietic depressors of hepcidin (or demonstrating that depressors have an effect only with concomitant transferrin desaturation).
- •
Inflammation sensing mediated through IL-6/STATs pathway and other cytokines upregulates hepcidin synthesis, being the key mechanism in hypoferremia of acute inflammation through the action of ferroportin degradation in macrophages.
Erythropoietic drive overrides both iron sensing and inflammation sensing mechanisms, but the extent to which these potentially opposing regulators of hepcidin synthesis play out in the context of iron metabolism in TIO is difficult to predict from murine studies alone and require careful clinical observations. These are discussed later under the clinical condition in question.
Cellular Iron Homeostasis
Intracellular iron homeostasis is controlled not only by the synthesis of ferritin but also by the regulation of iron uptake through regulation of membrane transferrin receptors (TfR). Both of these are regulated by IREs, stem-loop structures present in untranslated regions (UTR) of mRNA, for example, in the 5′UTR of H-ferritin or the 3′UTR of TfR mRNA, respectively. Both of these are sensitive to the magnitude of labile intracellular iron pools (LIP) through interaction with cellular IRPs; the conformation of IRPs and their binding to IREs are sensitive to LIP concentrations. IRE binding of both IRP1 and IRP2 increases in iron-deficient conditions, but both are rapidly degraded by iron and heme. IRP2 has predominant control overall, whereas IRP1 can switch from aconitase activity form in iron repletion (dependent on iron-sulphur cluster assembly, 4Fe-4S) to IRE-binding form in iron deficiency (losing iron: 3Fe-4S). Therefore, both their cellular level and the position of IRE on mRNA regulate in concert the onset and degree of translation events (5′UTR governing access to matrices, 3′UTR governing stability of matrices by regulating the binding of nucleases). High levels of LIP thus increase ferritin synthesis while decreasing the membrane expression of TfR1. However, in the erythron such feedback is absent; instead transcriptional control permits high TfR despite high cellular iron or heme, consistent with hemoglobin synthesis requirements. Most ferroportin transcripts also contain IRE at 5′UTR, and therefore the amount of mRNA is increased in iron overload. However, the effective regulation of ferroportin happens post-translationally through hepcidin-dependent down-regulation or lack of Fe(II) acceptor.
The availability of iron for the synthesis of iron-containing molecules at a cellular level is directed through a transient low molecular weight iron pool, LIP, which in turn determines the levels and action of IRPs. Although LIP iron has been proposed to be coordinated mainly by glutathione from a thermodynamic perspective, its exact nature still remains unclear, but it can potentially redox cycle between Fe(II) and Fe(III) with consequent generation of harmful free radicals. To minimize these risks, elegant homeostatic mechanisms carefully coordinate the distribution of body iron so as to provide iron pools for efficient synthesis of these proteins, while minimizing iron-mediated free radical generation.
Iron homeostatic mechanisms
Iron homeostatic mechanisms are key to the pathophysiology of transfusional iron overload (TIO). In humans, these mechanisms are best adapted to increasing iron acquisition in conditions of iron deficiency or anemia, or to limiting iron distribution from the macrophage system during inflammation. They are not well adapted, however, to controlling the distribution of TIO or to eliminating excess iron. This is in marked contrast to rodents, where most studies on iron overload and iron metabolism have been performed and where iron overload is eliminated efficiently by the biliary route. Iron homeostasis is adapted to supplying only that which is essential for the functioning of proteins involved in oxygen transport, oxidative energy production, mitochondrial respiration, and DNA synthesis, while minimizing the potential for iron toxicity from its redox cycling. These homeostatic mechanisms work at 2 levels: firstly at the level of the whole body through interactions of plasma hepcidin with membrane ferroportin and secondly at a cellular level through interaction of iron responsive element (IRE)-binding proteins (IRPs) with IREs present on mRNAs of key iron metabolism–related proteins.
Body Iron Homeostasis
A healthy human contains 40 to 50 mg/kg of iron, mainly as hemoglobin (30 mg/kg). About 4 mg/kg is present in muscle myoglobin, with 2 mg/kg in cells as iron-containing enzymes. Storage iron, present as ferritin and its compact, partially degraded form hemosiderin, ranges from 0 to 2000 mg ; this is mainly present in liver, spleen, and bone marrow (BM) macrophages, formerly referred to as the reticuloendothelial system (RES), and in hepatocytes. Liver iron concentration (LIC) rarely exceeds 1.8 mg/g dry weight (dw) in healthy individuals in the absence of liver disease, hemochromatosis genes, inappropriate dietary supplementation, or blood transfusion.
A healthy individual absorbs only about 10% of dietary iron or about 1 to 2 mg/d, usually balanced by iron loss from skin, gut, menstruation, or pregnancy. Anemia, hypoxia, ineffective erythropoiesis (IE), and the presence of variant HFE genes increase iron absorption, the common factor being low, or inappropriately low, plasma hepcidin levels. The latter permits higher enterocyte ferroportin expression, allowing Fe(II) flux and hence increasing dietary iron absorption. Iron absorption is also increased through hypoxia-inducible factor 1–mediated signaling, by duodenal upregulation of DcytB and DMT1 expression. Thus, in principle, any anemia will tend to increase the efficiency of iron absorption. Most body iron turnover, however, is not directed through iron absorption, but through plasma transferrin, which, although binding only 1 to 2 mg of iron at any moment, in a healthy adult delivers about 20 to 30 mg/d via transferrin receptors on the erythron for hemoglobin synthesis.
Hepcidin regulation is important both to iron absorption from diet and to iron egress from erythrophagocytic macrophages. Hepcidin controls iron egress from both macrophages and enterocytes by binding to and degrading ferroportin, through which Fe(II) exits these cells. Hepatic hepcidin synthesis is controlled by at least 3 distinct regulatory mechanisms responsive to levels of iron, erythropoiesis, or inflammation.
- •
Extracellular iron sensing involves the binding of diferric transferrin to transferrin receptor 1 (TfR1), initiating the translocation of HFE from TfR1 to TfR2 and its subsequent signaling via ERK1/ERK2 and p38 MAP kinase to induce hepcidin expression. Storage iron sensing is affected by BMP6 signaling via BMP receptor (and SMADs pathway) whose sensitivity is markedly increased by its interaction with hemojuvelin, HFE, and TfR2 in holotransferrin-dependent manner, thus enhancing hepcidin transcription.
- •
Erythropoiesis sensing involves BM-derived factors that suppress hepcidin synthesis; conditions with high levels of IE will have high levels of these factors, the nature of which has been debated. These include GDF15 ; twisted gastrulation factor-1 ; and most recently erythoferrone, which has been identified as a key factor in mice, although its relevance in humans has yet to be demonstrated. Another separate erythropoiesis sensing mechanism likely involves desaturation of transferrin by the erythron as disruption of transferrin iron uptake into erythron in hbd mouse increases hepcidin level despite ongoing anemia, presumably overriding the erythropoietic depressors of hepcidin (or demonstrating that depressors have an effect only with concomitant transferrin desaturation).
- •
Inflammation sensing mediated through IL-6/STATs pathway and other cytokines upregulates hepcidin synthesis, being the key mechanism in hypoferremia of acute inflammation through the action of ferroportin degradation in macrophages.
Erythropoietic drive overrides both iron sensing and inflammation sensing mechanisms, but the extent to which these potentially opposing regulators of hepcidin synthesis play out in the context of iron metabolism in TIO is difficult to predict from murine studies alone and require careful clinical observations. These are discussed later under the clinical condition in question.
Cellular Iron Homeostasis
Intracellular iron homeostasis is controlled not only by the synthesis of ferritin but also by the regulation of iron uptake through regulation of membrane transferrin receptors (TfR). Both of these are regulated by IREs, stem-loop structures present in untranslated regions (UTR) of mRNA, for example, in the 5′UTR of H-ferritin or the 3′UTR of TfR mRNA, respectively. Both of these are sensitive to the magnitude of labile intracellular iron pools (LIP) through interaction with cellular IRPs; the conformation of IRPs and their binding to IREs are sensitive to LIP concentrations. IRE binding of both IRP1 and IRP2 increases in iron-deficient conditions, but both are rapidly degraded by iron and heme. IRP2 has predominant control overall, whereas IRP1 can switch from aconitase activity form in iron repletion (dependent on iron-sulphur cluster assembly, 4Fe-4S) to IRE-binding form in iron deficiency (losing iron: 3Fe-4S). Therefore, both their cellular level and the position of IRE on mRNA regulate in concert the onset and degree of translation events (5′UTR governing access to matrices, 3′UTR governing stability of matrices by regulating the binding of nucleases). High levels of LIP thus increase ferritin synthesis while decreasing the membrane expression of TfR1. However, in the erythron such feedback is absent; instead transcriptional control permits high TfR despite high cellular iron or heme, consistent with hemoglobin synthesis requirements. Most ferroportin transcripts also contain IRE at 5′UTR, and therefore the amount of mRNA is increased in iron overload. However, the effective regulation of ferroportin happens post-translationally through hepcidin-dependent down-regulation or lack of Fe(II) acceptor.
The availability of iron for the synthesis of iron-containing molecules at a cellular level is directed through a transient low molecular weight iron pool, LIP, which in turn determines the levels and action of IRPs. Although LIP iron has been proposed to be coordinated mainly by glutathione from a thermodynamic perspective, its exact nature still remains unclear, but it can potentially redox cycle between Fe(II) and Fe(III) with consequent generation of harmful free radicals. To minimize these risks, elegant homeostatic mechanisms carefully coordinate the distribution of body iron so as to provide iron pools for efficient synthesis of these proteins, while minimizing iron-mediated free radical generation.
Impact of blood transfusion on iron balance
The rates and nature of blood transfusion regimens affect iron accumulation and its distribution in the body. This is key to the pathophysiology of iron overload and varies with the underlying clinical condition.
Thalassemia Major
In thalassemia major (TM), blood transfusion typically begins in the first year of life. Current transfusion recommendations in TM aim to keep the pretransfusion hemoglobin level at approximately 9.5 g/dL and to maintain an average hemoglobin of 12 g/dL, which usually amounts to an iron load rate (ILR) of 0.3 to 0.5 mg/kg/d. This regimen has been arrived at so as to balance the beneficial effects of suppression of IE and dietary iron absorption with the iron accumulated from transfusion. The transfusional suppression of the endogenous BM activity can be assessed by monitoring circulating transferrin receptors, which show more suppression when the pretransfusion hemoglobin level exceeds 10 g/dL. Maintenance of a mean pretransfusion hemoglobin level of 9.4 g/dL versus 11.3 g/dL decreased net blood consumption and was associated with improved control of iron overload in Italian patients. This optimal balance may not be universal and may depend on the severity of thalassemia genotype. In the prechelation era, LICs of 40 mg/g dw were typically seen by 10 years of age. Failure to control these levels risks extrahepatic spread of iron (see later discussion).
Sickle Cell Disease
The age of commencing blood transfusion, transfusional ILR, and the nature of the transfusion regimen itself, all affect the rate and extent of iron overload in sickle cell disease (SCD) and often differ considerably from TM. Net iron accumulation from transfusion in SCD is slower than TM, firstly because of differences in transfusion practice between these conditions and secondly because SCD patients tend to be in negative iron balance in the absence of transfusion. In SCD, there is considerable intravascular hemolysis leading to iron loss via urine (as in PNH) and possibly bile. Urinary iron loss in SCD may reach as much as 15 mg/d (∼0.2 mg/kg/d, ie, comparable to average SCD transfusional ILR). Furthermore, the marrow is less expanded in SCD than in TM or NTDT, leading to less hepcidin suppression and less tendency for increased iron absorption. Under conditions of hypertransfusion, where synthesis of sickle hemoglobin (HbS) is suppressed, or under vigorous chronic automated exchange procedures, where the percentage of HbS is maintained at low levels, intravascular hemolysis will also be suppressed and thus the tendency to lose iron through this mechanism will be diminished.
Historically, blood transfusions were typically sporadic and given by simple transfusion or by some form of partial exchange procedure in response to acute episodes, which over a lifetime would lead to significant iron overload. Transfusion has been increasingly given to prevent primary and secondary stroke. This approach, together with a wider use of transfusion to prevent or treat other complications, such as chest syndrome, or in preparation for major surgery, puts an increasing proportion of patients at risk of TIO. In a large multicenter international study, where most patients received simple (60%) or exchange transfusions (20%), the mean ILR was 0.22 mg/kg/d, notably lower than in TM. Manual exchange procedures, where about one-third of the blood volume is exchanged, lead to ILR of about 40% of simple transfusions, as estimated from ferritin increments. With automated erythrocytapheresis, ILR was only 0.053 mg/kg/d with a target pretransfusion HbS less than 50% compared with 0.39 mg/kg/d for simple transfusion with a target HbS less than 30% and 0.29 mg/kg/d with a target HbS less than 50%.
Other Conditions
In other forms of TIO, the rates of ILR again vary considerably; for example, a mean of 0.4 mg/kg/d was found in transfusion-dependent Diamond-Blackfan anemia (DBA) patients with 0.28 mg/kg/d in myelodysplastic syndrome (MDS) patients in the same study. Patients who receive repeated myeloablative chemotherapy cycles for leukemias or lymphomas can accumulate more than 100 units of transfused blood or 20 g of excess body iron that will eventually require removal if long-term iron toxicity is to be avoided.
Mechanisms of iron toxicity in transfusional overload
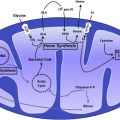
The pathophysiologic consequences of TIO, which are broadly observed in tissues in which storage iron accumulates at the highest concentrations, are summarized in Fig. 1 . Ferritin within cells is degraded in lysosomes or proteasomes, the iron is released into LIP, and this iron is reincorporated into new ferritin synthesis or made available for synthesis of essential iron-containing proteins. Once the LIP reaches a critical concentration, the iron can redox cycle between ferric Fe(III) and ferrous Fe(II) forms through the donation or acceptance of an electron and enhance the generation of reactive oxygen species (ROS), with a cascade of consequences (see Fig. 1 ). Both the concentration of LIP as well as the capacity of cells to accommodate increased levels of iron are likely to vary between cell type, and the exact nature and redox state of LIP remain unresolved. For example, in the human K562 cell line LIP concentrations of 0.24 to 0.4 μM have been estimated using the fluorochrome calcein as a probe. However, using electron paramagnetic resonance (EPR) spectroscopy, which detects Fe(III) and requires no manipulation of cells, an intracellular EPR-detectable high-spin ferric iron signal was found at approximately 3.2 μM. More recently, increased levels of LIP have been linked to increased ROS production and potentially oncogenic effects. Ferritin acts as a sink for LIP by decreasing its magnitude and its potential toxicity. For example, murine erythroleukemia cells overexpressing H-ferritin displayed lower levels of LIP and ROS.
Not all ROS are necessarily toxic to cells however. Large quantities of superoxide are produced naturally by respiration (about 30 g/d of superoxide) but their toxic potential is controlled by their conversion to water by superoxide dismutase and glutathione peroxidase. It is the favorable redox potential of the Fe(II)/Fe(III) couple (between +0.35 and −0.5 V) that allows it to redox cycle and thus catalyze the interaction of superoxide with hydrogen peroxide (H 2 O 2 ) through the Haber-Weiss reaction, generating highly reactive hydroxyl free radicals. The hydroxyl radical has a great affinity for electrons, will oxidize all substances within its immediate vicinity (diffusion radius of 2.3 nm), and has been shown to promote lipid peroxidation, with damage to organelles such as lysosomes and mitochondria. The interaction of the hydroxyl radical with lipid proceeds through the initial abstraction of a hydrogen atom (to yield a water molecule); molecular rearrangement of the lipid with peroxidation; and the formation of a peroxyl radical, which is able to propagate further lipid peroxidation in a chain reaction. The end result is decomposition of lipid molecules with concomitant effects on the integrity of organelles. Although organelle damage may lead directly to apoptotic cell death, this may also encourage fibrogenesis as iron-induced aldehyde lipid peroxidation products such as MDA and 4-HNE promote collagen gene expression. Fibrogenesis is also associated with autocrine production of TGFβ-1 in stellate cells (see Fig. 1 ). ROS also damage DNA, risking genomic instability, mutagenesis, and cell death or neoplasia. ROS also directly activate caspases, thereby accelerating apoptosis, but, paradoxically, may also have antiapoptotic effects by activating NF-kB, which may contribute to MDS transformation and to iron-mediated neoplasia, such as hepatoma.
An important, often neglected, mechanism of toxicity from iron overload is that of the increased risk of infection, which is the second commonest cause of death in TM. Several mechanisms come into play, the most important being transferrin saturation (TfS). This protein, in addition to its pivotal role in supplying iron to the erythron and other tissues, naturally exists where only an average of one-third of its 2 iron binding sites are occupied with Fe(III). A key role of transferrin is to deprive bacteria of the iron that these microorganisms require to grow. Although some bacteria have adapted to use transferrin iron, most have not, and so there is a paradigm shift in the availability of iron to microorganisms once transferrin becomes saturated. Other mechanisms, such as effects on neutrophil function have been postulated to be affected by TIO. Recent work has shown that following blood transfusion, NTBI is liberated from the rapid catabolism of a proportion of nonviable red cells. In principle, this and other forms of NTBI in plasma will be more available to microorganisms than transferrin iron.
Distribution and consequences of TIO
Iron Distribution and Consequences in Thalassemia Major
The impact of chronic blood transfusion on body iron distribution is most completely described in TM, where transfusion typically begins in the first year of life. Transfused iron initially accumulates as storage iron in spleen, liver, and BM macrophages and later in hepatocytes, with 80% of storage iron in the liver. The storage capacity of the macrophage system following blood transfusion has not been recently studied, but historical sources estimate it at about 10 g. Histologic descriptions using Perl’s stain show that with increasing TIO, increasing proportions are seen in hepatocytes once the macrophage system is saturated. Interestingly, particularly with optimal chelation therapy, TM patients today have low iron concentrations in hepatocytes, whereas macrophage iron remains present ; this contrasts with NTDT where iron accumulates through the portal system and concentrates in peri-portal hepatocytes with macrophage sparing. This distribution in NTDT is thought to be influenced further by low hepcidin levels, and therefore high macrophage ferroportin, due to high levels of IE typical in thalassemia.
As TIO evolves, particularly with suboptimal chelation therapy, a variable proportion of iron ‘escapes’ from the liver into the endocrine tissues and heart. This gives rise to the classic pathology, morbidity, and mortality historically associated with TIO. An understanding of the effects of blood transfusion on body iron distribution is best appreciated from post-mortem data obtained during prechelation era, because iron chelation fundamentally alters body iron distribution, being relatively tropic for hepatocellular iron compared with extrahepatic iron. Data obtained under these circumstances showed that in patients dying from complications of TIO, iron was unevenly distributed in the body, with high concentrations present in liver, heart, and endocrine tissues; very low in striated muscle; and none in the brain. Remarkably, these patients typically died of heart failure in the second and third decades of life, although the myocardial iron concentration (MIC) was a fraction of that in the liver. This observation has recently been supported by magnetic resonance imaging (MRI) evidence ; examination of myocardial tissue both biochemically and by MRI at post mortem in patients dying from iron-induced cardiomyopathy showed an average MIC of only 5.98 ± 2.42 mg/g dw. Evidently, the heart is less adapted to accommodating high concentrations of storage iron than the liver, even though the storage iron is not directly toxic to cells (see later discussion).
Before the introduction of cardiac MRI monitoring and newer chelation regimens, the frequency of heart failure, diabetes, hypothyroidism, and hypoparathyroidism were all falling. Hypogonadism (typically hypogonadotropic) is still an early and common feature of iron overload in TM, presenting with primary or secondary amenorrhea in women or poor growth and delayed puberty. Since the introduction of cardiac MRI imaging and the intensification of chelation therapy in selected patients with increased MIC (mT2*<20 ms), the incidence of heart failure has fallen further. Indeed, in a recent cohort analysis of patients observed for a decade by cardiac MRI and receiving individually tailored chelation, heart failure was no longer the leading cause of death and the proportion of patients with myocardial iron (mT2*<20 ms) fell from 60% to 23%. Cirrhosis, which develops 1 or 2 decades after heart failure, is becoming more common as patients live longer, being present in about 50% of patients at post mortem and is particularly common in patients with chronic hepatitis. Similarly, hepatocellular carcinoma is becoming more common.
The relationship between the accumulation of liver iron and the risk of extrahepatic spread has been the source of intense debate. Early work suggested a close relationship in TM between the control of LIC with deferoxamine and long-term outcome from cardiomyopathy. Post-mortem data in other diseases in the absence of chelation also suggested a relationship between transfusional ILR, LIC, and MIC. When cardiac T2* became available in patients receiving a variety of chelation regimens, only a weak correlation between LIC and mT2* was seen, and it was argued therefore that control of LIC was not important to controlling MIC in TM and therefore to limiting potentially fatal cardiomyopathy. The interpretation of the UCLH group was that this lack of correlation was mainly due to the high proportion of patients in this study having been on intensive chelation therapy with desferoxamine, which was subsequently shown to decrease LIC at a faster rate than myocardial iron ; this would therefore mask a potentially important relationship between these variables. Noetzli and colleagues have somewhat clarified this issue by demonstrating the importance of longitudinal rather than cross-sectional analysis of the relationship. They showed that failure to control LIC over several years with chelation increased the risk of myocardial iron deposition. Conversely, LIC reduction had a delayed effect on decreasing the MIC. Longitudinal UK studies of the LVEF relationship to mT2* show that the risk of heart failure increases for mT2* less than 10 ms, which approximates to MIC greater than 2.7 mg/g dw. It can be concluded that LIC control in TM is important both to limiting liver damage and to controlling MIC, thus markedly reducing the risk of iron-mediated cardiomyopathy with heart failure.
Iron Distribution and Consequences in SCD
In patients receiving sufficient repeated transfusions to cause TIO, clinical consequences begin later than in TM, and thus effects on growth and sexual development are relatively uncommon. With transfusion, iron from erythrocyte catabolism initially accumulates in macrophages (Kupfer cells, sinusoidal compartment), but later, when the LIC exceeds 7 mg/g dw, in hepatocytes (based on the Angelucci formula, being equivalent to about 5 g of transfused iron in a 70 kg adult, or about 25 units of transfused blood). Hepatocellular iron stores in SCD only approached those of the sinusoidal compartment when total liver iron levels were high (>15 mg/g dw or about 50 units of transfused red cells). Accumulation of liver iron without adequate chelation therapy risks fibrosis and cirrhosis. Fibrosis has been reported as early as 2 years after initiation of transfusion and in about one-third of patients with LIC greater than 9 mg/g dw and in direct proportion to the LIC, correlating with LIC in the absence of hepatitis C infection. The true frequency of cirrhosis in multitransfused adult SCD patients is not clear. Post-mortem studies found cirrhosis in 11% of all patients and in nearly half of patients who died with severe liver siderosis.
The extrahepatic consequences of iron overload, particularly endocrine and cardiac effects, appear to be later or more delayed in SCD than TM. Myocardial iron deposition, as judged by T2* of less than 20 ms, is rare and after more transfusion episodes in SCD than TM. However, post-mortem studies show iron deposition in the heart in heavily transfused patients. In SCD and TM patients matched for LIC, the incidence of heart disease, gonadal failure, and endocrine disturbances including growth delay, appear to be less for age less than 20 years in SCD. Despite these differences from TM, SCD patients are unlikely to be completely protected from the extrahepatic effects of TIO and indeed cases of myocardial iron are reported by MRI. Recent preliminary studies from the Multi Center Study of Iron Overload (MCSIO) group found evidence of increased pituitary iron in SCD with highest LIC. In one study, patients with the lowest bone mass also had the highest serum iron values, although serum ferritin (SF) was within normal limits in these patients. Furthermore there was an inverse correlation between the estimated pituitary iron and the pituitary volume, and hence its endocrine reserve. MRI may, in principle, identify early pituitary iron deposition in SCD before clinical manifestations are apparent.
Increased iron has also been identified by MRI in the kidney. This signal is highest in nontransfused patients with high levels of LDH, lacks correlation with LIC, and is higher than in TM patients, suggesting it originates from iron taken up by kidney from hemoglobin freed during intravascular hemolysis rather than iron delivered by transferrin or NTBI as a consequence of TIO. This signal also suggests that kidney R2* may be a biomarker for chronic hemolysis-mediated vascular complications in SCD. The extent to which this mechanism is implicated in renal damage in SCD is not clear.
Iron Distribution in Other Forms of TIO
TIO is seen in an increasing number of underlying conditions ( Table 1 ). Iron accumulation in MDS may start even before patients become transfusion-dependent because of IE, which although variably counterbalanced by increased levels of cytokines upregulated during infections (eg, IL-6), may still inhibit hepcidin production with subsequent increased iron absorption from the gut, as described earlier. Once transfusion begins, as with other forms of TIO, iron initially accumulates in RES and then in the liver. A key question with respect to the pathophysiology of iron overload is how rapidly iron spreads extrahepatically and how rapidly this is likely to be problematic and hence require chelation treatment. In the prechelation era, patients examined at post-mortem had an increased risk of myocardial iron with increasing numbers of blood transfusions. Early studies with cardiac MRI showed increasing risk of myocardial iron as the number of transfusions exceeded 50 units. Analysis of the relationship between transfusion and myocardial iron is complicated because patients have received chelation therapy in some but not all reports. Overall, in nonchelated MDS patients, iron spread to myocardium occurs after approximately 70 to 100 units of blood (containing 14–20 g iron).
Related posts:



Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


