Fig. 25.1
The incidence of MDS and age. Age adjusted incidence/100,000 individuals. Age in five block years. (Data from Rollison DE, Howlader N, Smith MT et al. Epidemiology of myelodysplastic syndromes and chronic myeloproliferative disorders in the United States, 2001–2004, using data from the NAACCR and SEER programs. Blood 2008;112:45.)
MDS is characterized by dysplastic, ineffective hematopoiesis. The bone marrow is typically hypercellular for the patient’s age with peripheral cytopenias and an increase in hematopoietic precursors in the bone marrow and/or peripheral blood. Apoptosis, programmed cell death, is increased in MDS resulting in an increased rate of progenitor cell division but with an impaired production and release of mature cells [12, 13]. The clonal origin of MDS has been confirmed by isozyme analysis of glucose 6 phosphate dehydrogenase (G6PD) in heterozygous females and more recently by molecular analysis of other loci such as the androgen receptor gene [14, 15]. The involvement of lymphoid progenitors is more variable and it remains controversial if B and T lymphocytes are also part of the abnormal dysplastic clone [16, 17]. The appearance of a clonal abnormality appears to be an early event and cytogenetic abnormalities can be demonstrated before any morphological or clinical findings of MDS [15, 16]. The initial cytogenetic events are believed to be part of a multistep process that predisposes the pluripotential stem cell to secondary genetic events [18]. Alterations in one or more oncogenes including N-RAS, p53, IRF-1 tumor-suppressor genes, methylation of specific gene promoters, and transcription factor genes have all been associated with the stepwise progression of MDS [19, 20]. The progression of MDS is frequently characterized by a progressive increase in chromosomal instability that leads to the development of aberrant clones and the development of complex karyotypes. The mechanisms responsible for the chromosomal instability are not known. Telomeres are thought to be critical in the maintaining normal hematopoiesis and are postulated to play a role in the progressive chromosomal instability in MDS [21]. Telomeres are noncoding repeated sequences at chromosome ends that function to stabilize chromosomes and prevent chromosomal aberrations. Each somatic cell division is associated with loss of telomere length. The cumulative effects of telomere shortening lead to cell senescence and is part of the normal aging process [22]. A reduction of telomere length is observed in a subset of MDS patients. The shortening of telomeres is noted in patients with progressive, advanced MDS with multiple complex karyotypic abnormalities and the genetic instability associated with shortening of telomeres may contribute, in part, to the leukemic transformation in some patients with MDS [23, 24]. Alteration of telomere dynamics in hematopoietic cells may precede the development of MDS and result in genetic instability and predispose to the development of MDS and AML. However, normal hematopoietic cells also express varying levels of telomere shortening depending on their degree of differentiation and activation [22].
The hematopoietic microenvironment may also play a role in the pathophysiology of MDS [25]. The bone marrow stroma responds to signals from the hematopoietic cells and is abnormal in some patients with MDS. Abnormalities of the bone marrow microenvironment may affect apoptosis and telomere shortening in clonal hematopoietic cells [26].
Genetic, environmental, and exposure factors have been associated with an increased risk for the development of MDS [27, 28]. Inherited constitutional genetic defects have been associated with up to 30 % of children with MDS and related myeloproliferative disorders [29–32]. Children with Shwachman–Diamond syndrome, Fanconi anemia, dyskeratosis congenital, and neurofibromatosis type 1 have constitutional genetic defects that are associated with the increased risk for the development of both MDS and AML [33, 34].
Mutations of specific genes mediating DNA repair appear to predispose to the acquisition of secondary cytogenetic abnormalities that can lead to the development of MDS [35]. Somatic mutations are common in patients with MDS occurring in 51 % of patients and may be associated with specific clinical features [36]. These point mutations correlated with the clinical phenotype, specific cytopenias, disease progression, and overall survival. Genes encoding runt-related transcription factor 1(RUNX1), tumor protein p53(TP53), and neruroblastoma RAS viral oncogene homologue (NRAS) were associated with thrombocypoenia and an increased percent of blast forms [36]. Point mutations resulting in the activation of the specific genes (TP53, EZH2, ETV6, RUNX1, and ASXL1) were independent markers of poor prognosis and may, in part, explain the clinical heterogeneity of MDS. TET2 mutations was the most common genetic abnormality but was not associated with clinical progression. TET2 mutations are not unique to MDS and are found in a number of myeloid neoplasms and occurred across a spectrum cytogenetic subgroups and other somatic mutations suggesting that TET2 mutations have a pathogenic role that may be independent of other abnormalities [35]. Mutations of NRAS have been reported in patients with MDS and are associated with chromosomal instability and disease progression [32 33]. The frequency of these mutations increases with progression of the disease and the subsequent development of secondary cytogenetic events and AML [33]. The etiological role of each of the somatic point mutations in the development of and progression of MDS is unclear and these mutations may just reflect the genetic instability of the abnormal clone and its propensity to develop random genetic mutations [34]. MDS patients have defects in a number of signal transduction pathways that appear to be related to the evolving ineffective hematopoiesis and epigenetic changes [35, 36]. These acquired abnormalities, however, may contribute to the further dysregulation of progenitor cell cycle kinetics, response to cytokines, and the maintenance of DNA integrity, which results in progressive genetic instability [37]. Abnormal regulation of microRNAs (miRNAs) which function as epigenetic regulators of gene expression may play a role in MDS. Alterations in miRNAs may be involved in the pathogenesis and may be independent markers of prognosis [37]. Abnormalities of the bone marrow microenvironment and aberrant cytokine production including abnormalities in the regulation of tumor necrosis factor alpha (TNF-a), transforming factor beta, and interleukin 1b which and may, in part, support the continued growth and expansion of the abnormal MDS clone [38–40].
The bone marrow in MDS has an increase in the number of apoptotic cells and the accelerated apoptosis is most marked in the less proliferative, better prognostic, forms of MDS [41, 42]. The etiological role of apoptosis in MDS, however, remains controversial. The resistance of precursor MDS cells to apoptosis could be one of the factors that confers a survival advantage to the MDS clone and is a common histopathologic finding that correlates with the ineffective hematopoiesis [43]. The lineage committed MDS cells, however, do appear to be more susceptible to apoptosis as they differentiate, which may reflect their disordered, ineffective, and impaired maturation [42]. The increased apoptosis of committed precursors could result in the ineffective and dysplastic maturation and the observed peripheral cytopenias that is discordant with the increased bone marrow cellularity and increased proliferation of immature precursors [44]. The increased apoptosis may also be a secondary event reflecting the impaired maturation and proliferation of the abnormal cone modulated by changes in the bone marrow microenvironment [43, 44]. The overexpression of TNF-a produced by MDS mononuclear cells can inhibit the growth of residual normal hematopoiesis and lead to increased cell death of normal precursors and a growth advantage of the abnormal MDS precursors [44].
MDS is associated with a number of immunoregulatory abnormalities including the development of autoantibodies and monoclonal gammopathies [45]. In subsets of patients with MDS, autoreactive T-cell clones are present that inhibit autologous erythroid and granulocytic colony growth [46, 47]. T-cell-mediated suppression of bone marrow growth and maturation appears to be an important development of the hypoplastic variants of MDS, as in patients with aplastic anemia [47, 48]. The immunoregulatory abnormalities may explain the response to immunosuppressive therapy in selected patients with MDS [49].
Environmental agents have been implicated in the etiology of MDS. In case-controlled studies, there is an association between MDS and cigarette smoking, exposure to benzene, petroleum products, organic solvents, fertilizers, pesticides, and inorganic dusts [50–54]. There appear to be defined genetic predisposing factors in some MDS patients that relate to naturally occurring complex DNA polymorphisms in genes that mediate DNA repair and the metabolism of environmental carcinogens [55, 56].
In selected genetically predisposed individuals, MDS may arise as a result of cumulative environmental exposures and studies have linked the development of MDS and the nonfunction 609 C.T polymorphic allele of the NAD(P)H:quinone oxidoreductase (NQO1) gene [55]. These genes appear to play a critical role in detoxifying benzene and its metabolites. This association is controversial, but may explain the increased incidence of MDS in some patients exposed to organic solvents and benzene-containing compounds [57, 58]. Similar controversial, but provocative results have been noted in the glutathione S-transferase (GST) genes that mediate the metabolism of cytotoxic and genotoxic agents [58, 59].
A prior exposure to alkylating agents (i.e., cyclophosphamide, chlorambucil, melphalan, etc.) is associated with an increased risk of MDS. Therapy-related MDS (t-MDS) represents approximately 10–20 % of MDSs and MDS/myeloproliferative disorders [60] (Table 25.1). The risk is, in part, related to the dose and duration of the cytotoxic therapy and generally occurs 3–7 years after the exposure [61]. Patients who received combination radiation therapy and chemotherapy are at greater risk for the development of t-MDS. Patients with Hodgkin’s lymphoma treated with alkylating agent-based chemotherapy regimens and radiation therapy have a 13 % projected incidence of t-MDS at 10 years posttreatment [61]. Total body irradiation, administered as part of the preparative regimen for an autologous stem cell transplantation, is associated with an increased risk for MDS, and the combination of high-dose alkylator therapy and total body irradiation was associated with a 10–15 % risk of t-MDS and secondary AML [62].
Table 25.1
Cytotoxic drugs implicated in the development of MDS
Class/drug |
|---|
Alkylating agents Busulfan Carboplatin Carmustine Chlorambucil Cisplatin Cyclophosphamide Dacarbazine Dihydroxybusulfan Lomustine Mechlorethamine Melphalan Mitomycin C Procarbazine Semustine Thiotepa |
Nucleoside analogs Fludarabine 2-Chlorodexoyadenosine (Clardribine) |
Antimetabolites 6-Mercaptopurine Methotexate Azathioprine |
Radiotherapy Usually involving large fields, e.g., TBI, large field therapeutic irradiation immunomodulator |
The chronic administration of purine analogs and antimetabolites has been associated with the development of MDS and AML [63, 64]. The MDS that occurs after chemotherapy exposure has a very poor prognosis [63, 64]. Therapy-related MDS is associated with a high frequency of karyotypic abnormalities involving chromosomes 5 and/or 7 and is characterized by complex karyotypes with the loss of genetic material in the form of deletions of all or part of these chromosomes [65]. In contrast to AML, balanced cytogenetic abnormalities including translocations and inversions are rare in MDS. The cytogenetic abnormalities in MDS are independent prognostic risk factors for overall survival and risk of the development of AML [65] (Table 25.2). Complex karyotypes (>3 abnormalities) typically include abnormalities of chromosomes 5 {−5/del (5q)} and/or 7 {−7/del (7q)} are associated with an unfavorable clinical course and a poor prognosis. Cytogenetic abnormalities also are associated with characteristic morphologic abnormalities such as isolated del(20q) with involvement of erythroid cells and megakaryocytes and development of thrombocytopenia. However, in most patients there is no history of exposure to known mutagens, cytotoxic agents, or environmental agents and therefore the etiology for most patients with MDS is idiopathic or unknown.
Table 25.2
Cytogenetics in MDS and risk stratification system
Risk group | Karyotypes | Median survival, months | Time to 25 % of patient developed AML—months |
|---|---|---|---|
Favorable | 5q-,12p-,20q-,+21,-Y,11q t(11(q23)), normal any 2 abnormalities including 5q- | 51 | 71.9 |
Intermediate-1 | +1q,3q21/q26 abnormalities, +8,t(7q),+19, −21, any other single abnormality, any double abnormality not including abnormalities of chromosomes 5q or 7 | 29 | 16 |
Intermediate- 2 | −x,−7 or 7q,any double abnormality with −7 or 7q, complex with three abnormalities | 15.6 | 6 |
Unfavorable | Complex with >3 abnormalities | 5.9 | 2.8 |
Classification
The classification and diagnosis of MDS is evolving to incorporate new cytogenetic and molecular markers used in the diagnosis of myeloid malignancies. The updated WHO classification addresses the heterogeneity of the syndromes and attempts to separate MDS from reactive processes and other malignant stem cell disorders. The classification systems are dynamic and at times conflicting and clinically inconsistent reflecting differences in our understanding of the etiology, pathogenesis, and prognostic variables in MDS. The FAB (French–American–British) group was the first to define morphological criteria in the blood and bone marrow for the diagnosis and classification of MDS [66]. The FAB classification was based solely on morphology, and the percentage of blast forms in the blood and bone marrow (Table 25.3). The FAB classification was useful in categorizing patients with MDS, separating MDS from AML and other myeloproliferative disorders, and broadly assessing prognosis. This classification system, although generally adopted at the time, was clinically and biologically inconsistent [67]. The separation of MDS from AML and other clonal disorders was based on an arbitrary number of blast forms. Moreover, many patients with MDS had clinical and laboratory features of AML, aplastic anemia, and myeloproliferative disorders which were not addressed in the FAB classification [68, 69]. The FAB group subgroups also did not address the clinically important cytogenetic changes in MDS and were too variable to accurately predict prognosis, survival, or transformation to AML. The FAB classification, however, remained as a widely accepted classification system for diagnosis of MDS for two decades. The FAB group defined five categories: subtypes of MDS based on morphologic dysplasia, cytochemical stains for iron to detect ringed sideroblasts, percentage monocytes, and the percentage of blast forms in the bone marrow and peripheral blood. The FAB group classification included five categories: refractory anemia (RA) or refractory cytopenia; refractory anemia with ringed sideroblasts (RAEB); refractory anemia with excess blasts (RAEB); chronic myelomonocytic leukemia (CMML); and refractory anemia with excess blasts in transformation (RAEB-T).
Table 25.3
The French–American–British (FAB) classification of the Myelodyplastic Syndromes
Category | Peripheral blood | Bone marrow | |
|---|---|---|---|
Refractory anaemia (RA) or refractory blasts ≤ 1 %. Cytopeniaa | Anaemia,a blasts ≤ 1 % | AND | Blasts <5 %, ringed sideroblasts ≤15 % |
Refractory anaemia with ringed sideroblasts (RARS) | Anaemia, blasts > 1 % | AND | Blasts <5 % ringed sideroblasts > 15 % of erythroblasts |
Refractory anaemia With excess of blasts (RAEB) | Anaemia, blasts > 1 % blasts < 5 % | OR AND | Blasts ≥ 5 % BUT Blasts ≤ 20 % |
Chronic Myelomonocytic Leukemia (CMML) | Monocytes > 1 × 109/l | Blasts up to 20 % promonocytes often increased | |
Refractory anaemia with excess of blasts in rods | Blasts ≥5 % blasts | OR Auer OR | Blasts > 20 % Blasts < 30 % transformation (RAEB-T) |
The FAB classification separated the MDSs from other clonal hematopoietic disorders and defined the characteristic morphologic features of MDS in the peripheral blood and bone marrow. The FAB classification defined MDS as a clonal stem cell disorder characterized by ineffective, dysplastic hematopoiesis with morphological dyplasia and an imbalance between proliferation and maturation. The diagnosis of MDS required morphological dysplasia in one or more the hematopoietic cell lines and dysplasia should be noted in >10 % of cells of the involved lineage in either the bone marrow or the blood (Table 25.4). The morphological dysplasias, however, are generally not specific or diagnostic of MDS, but some of the morphological abnormalities are more characteristic of MDS and are useful in confirming the diagnosis. The neutrophil and megakaryocytic changes are the most specific and characteristic features in MDS [68, 70]. The presence in the peripheral blood of the acquired, pseudo, Pelger–Huet anomaly is a frequent and useful characteristic finding in the MDSs. This acquired abnormality resembles the inherited Pelger–Huet anomaly, therefore the designation of pseudo and is characterized by mature neutrophils that are hypolobulated with a single lobe or two joined by a thin band of chromatin, or dumbbell- or peanut-shaped bilobed nucleus [71]. The finding of dysplastic megakaryocytes in the bone marrow is also more suggestive of MDS than a reactive process [72–74]. In the peripheral blood, the platelet count may be low, normal, or increased with giant dysplastic platelets including hypogranular and agranular platelets. In the bone marrow, megakaryocytes may have bizarre nuclear shapes, with multiple separate nuclei and poor cyoplasmic granulation. The finding of micromegakaryocytes and megakaryocytes that are the size of a myeloblast with one or two abnormal small nuclei in the bone marrow is one of the most characteristic and recognizable morphological features in MDS. The megarkaryocytes may also demonstrate decreased or absent lobulations with separate and distinct nuclei. The FAB group noted that the majority of patients with MDS presented with a macrocytic anemia with a low reticulocyte count with a hypercellular bone marrow for the age of the patient. Red cell abnormalities include anisocytosis (abnormalities in size), poikilocytosis (abnormalities in the shape) including target cells, elliptocytes, stomatocytes, red cell fragments, basophilic stippled red cells (erythrocytes that contain aggregates of ribosomal RNA in the cytoplasm), and Pappenheimer bodies (red cells that contain basophilic iron-containing granules) may be noted on the peripheral blood film. In the bone marrow erythroid finding is common and include nuclear budding and internuclear bridging. Megaloblastic erythroid changes are common and are characterized by nuclear cytoplasm asynchrony, in which the nucleus is enlarged with open chromatin and less mature that is the cytoplasm. Ring sideroblasts are noted on the iron stain. Ring sideroblasts are erythroid precursors, erythroblasts, with iron staining in the mitochondria (>5 ferritin granules) forming a necklace at least a third or more around the nucleus. The FAB group proposed that ringed sideroblasts should account for more than 15 % of all nucleated cells in the bone marrow, but in the WHO classification ring sideroblasts should be >15 % of erythroid precursors [68, 75]. There is also an increase in sideroblasts in which iron-containing granules are increased in size and number but do not form a ring. Iron stores are generally increased when a patient presents even in the absence of transfusions [73]. Abnormal granulopoiesis is usually evident on the peripheral smear and includes neutropenia, hypersegmented neutrophils, with decreased or absent cytoplasmic granules. Myeloblasts may be noted on the peripheral smear and may contain Auer rods, thin clumps of azurophilic granules in the cytoplasm that are aggregates of fused lysosomes (Fig. 25.2). The bone marrow biopsy may reveal nonparatrabecular cluster of immature forms that stain with anti-CD34 antibodies (ALIP) [75]. The FAB group acknowledged that the distinction between MDS, AML, and myeloproliferative disorders can be difficult. Myeloid precursors are abnormal and the differentiating and recognition of blasts from dysplastic promyelocyts and myelocyte while critical in the diagnosis of AML versus MDS can be problematic [68].
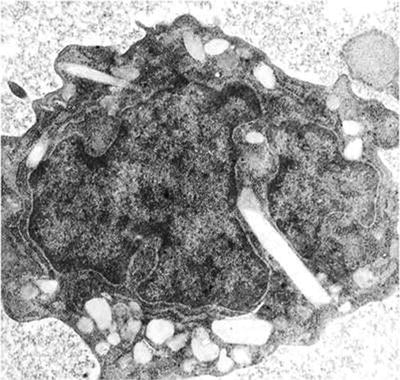
Table 25.4
Morphologic features of dysplasia
Lineage | Peripheral blood | Bone marrow |
|---|---|---|
Erythroid | Macrocytosis | Megaloblastic changes |
Elliptocytes | Nuclear budding | |
Acanthocytes | Ringed sideroblasts | |
Stomatocytes | Nuclear fragments | |
Teardrops | Cytoplasmic vacuolization | |
Basophilic stippling | Multinucleation | |
Acquired thalassemia | ||
Myeloid | Pseudo-Pelger-Huet anomaly | Abnormal Maturation Increase in monocytoid forms Abnormal localization of immature precursor (AILP) Hypo granulation Increased blasts |
Ayer rods | ||
Hypo granulation | ||
Hypersegmentation | ||
Ring shaped nuclei-circle | ||
Cell blasts forms | ||
Megakaryocyte | Giant platelets | Micromegakaryocyte |
Hypogranular or agranular | Hypo granulation | |
Platelets | Multiple small nuclei | |
Thrombocytopenia | Nuclear hypo lobation | |
Thrombocytosis |
Fig. 25.2
An electron micrograph of a myeloblast containing an Auer rod (Reprinted with permission from Lowenberg B. New Engl J Med 2007; 367:2208. From the collection of Dr. Janine André-Schwartz.)
The World Health Organization (WHO), in collaboration with the Society for Hematopathology and the European Association of Hematopathology in 2001, proposed a revision of the FAB morphological approach to the classification of MDS [76]. The WHO classification was updated in 2008 [77] (Table 25.5). The WHO classification, similar to the FAB classification, is based in part on the number of blasts in the peripheral blood or bone marrow. The WHO classification attempts to combine clinical, morphologic, immunophenotypic, and genetic features to define clinically important and useful subtypes. The WHO classification stratified the myeloid neoplasms into disorders comprised mainly blasts with minimal on no differentiation (AMLs) and neoplasms with either effective maturation (myeloproliferative neoplasms) or ineffective maturation with dysplastic features (MDS). Disorders that demonstrated both ineffective and effective maturation were classified as myelodysplastic/myeloproiferative neoplasms (MDS/MPN) [78] (Table 25.6). Moreover, if the clinical setting is the primary event that determines the myeloid neoplasm, then the classification of the disorder should reflect its unique biologic factor, i.e., therapy-related MDS (t-MDS). The WHO classification has supplanted the FAB classification for myeloid neoplasms. The WHO classification included requirements for the type of specimens to be obtained, the assessment of blasts, assessment of blast lineage, and cytogenetic or mutational studies. The assessment of blasts in the peripheral blood (PB) and bone marrow (BM) should be obtained prior to any definitive therapy. The BM biopsy should be at least 1.5 cm in length and cytogenetic analysis and, when indicated, flow cytometry should be performed, with additional material cyropreserved for later molecular genetic studies as needed [78]. The WHO lowered the threshold for percent of blasts to diagnose AML from 30 % (FAB) to 20 %. The FAB diagnosis of RAEB-T was therefore eliminated and was reclassified as AML with multilineage dysplasia. The percent of blasts should be derived from a 200-cell leukocyte differential count of the PB smear and a 500-cell differential count of all nucleated BM cells. The blast percentage includes myeloblasts, monoblasts, promonocytes, and megakaryoblasts. Proerythroblasts are not counted as blasts except in the diagnosis of pure acute erythroleukemia. The use of flow cytometrics to determine the percent of blasts, assessment of CD34+ cells, was not recommended as a substitute for visual inspection unless the aspirate was of poor quality. The percent of CD34+ cells generally correlates with morphologic examination of routine BM and PB smear; however, the WHO noted that not all leukemic blasts express CD34 and hemodilution and processing artifacts can yield misleading results. Multiparameter flow cytometry was recommended to determine the blast lineage and to determine aberrant antigen expression. Immunophenotypic aberrancies are common in MDS and may aid in the distinction of clinically relevant subgroups in MDS which may be helpful in assessing prognosis and predicting response to treatment [79, 80]. However, it remains unclear if these changes are specific for the diagnosis of MDS as they occur in other myeloid neoplasms [79, 81]. Immunophenotyping combined with cytogenetics and morphology help in the diagnosis and assessing evolution of MDS to AML [80]. An increase in the percentage of CD34+ cells or CD117+ cells may provide further evidence of the transformation of a low-grade MDS. The WHO, however, while noting that aberrant antigen expression patterns for MDS have been noted did not consider the phenotypic abnormalities sufficient, in the absence of conclusive morphologic and/or cytogenetic abnormalities, for the diagnosis of MDS. The finding of three or more phenotypic abnormalities involving one or more of the myeloid lineages should be considering suggestive, but not diagnostic of MDS. Patients whose cells demonstrate aberrant immunophenotypic markers should be followed for morphologic features sufficient to diagnose MDS. The use of additional studies including FISH analysis and mutational analysis including JAK2, MPL, and KIT should be performed as clinically indicated [82]. FISH analysis is helpful in defining common cytogenetic abnormalities in MDS. The WHO removed CMML from the MDS classification and assigned it into a separate myelodysplastic/myeloproliferative neoplastic group (MDS/MPN) (Table 25.7). The myeloproliferative disorders were renamed in the WHO 2008 classification as myeloproliferative neoplasms (MPNs) reflecting their clonal neoplastic origins. The MPN are characterized by increased but effective hematopoiesis without prominent dysplasia. However, MPN can present with dysplastic changes in one or more lineage. The importance of diagnostic cytogenetic and somatic mutations such as JAK2 V617F and defined histopathologic features help in defining the MPN [81]. The myelodysplastic/myeloproliferative neoplasm (MDS/MPN) group is an overlap disorder reflecting that some patients have features of both MDS and MPN and patients present with a clinical picture that demonstrates both increased proliferation with maturation and dysplastic and ineffective maturation. The classification of MDS/MPN reflects difficulty separating MDS from other myeloid neoplastic disorders and the cytogenetics and molecular diagnostic tests that are useful in defining the myeloproliferative neoplasms.
Table 25.5
2008 WHO classification of myelodysplastic syndromes
Name | Peripheral blood key features | Bone marrow key features |
|---|---|---|
Refractory cytopenias With unilineage Dysplasia Dysplasia (RCUD) RA Anemia (RA) | <1 % blasts | Erythroid dysplasia |
Refractory neutropenia (RN) | Neutropenia <1 % blasts | Granulocytic dysplasia |
Refractory thrombocytopenia (RT) | Thrombocytopenia <1 % blasts | Megakaryocytic dysplasia |
Refractory anemia with ring sideroblasts RARS | Anemia no blasts | Unilineage erythroid dysplasia ≥15 % of erythroid precursors ring sideroblasts |
Refractory cytopenia with multilineage dysplasia RCMD | Cytopenia(s) <1 % blasts No Auer rods | Multilineage dysplasia ± ring sideroblasts |
Refractory anemia with excess blasts Type 1 RAEB-1 | Cytopenia(s) <5 % blasts No Auer rods | Unlineage or multilineage dysplasia 5 %–9 % blasts No Auer rods |
Refractory anemia with excess blasts Type 2 RAEB-2 | Cytopenia(s) 5 %–19 % blasts ± Auer rods | Unilineage or multilineage dysplasia 10 %–19 % blasts ± Auer rods |
MDS associated with isolated del(5q) Del(5q) | Anemia normal or high platelet count | Isolated 5q31 deletion hypolobated megakaryocytes |
Childhood MDS Including refractory Cytopenia of childhood (provisional) RCC | Pancytopenia | <5 % marrow blasts for RCC Marrow usually hypocellular |
MDS, unclassifiable MDS-U | Cytopenias ≤ 1 % blasts | Does not fit other categories Dysplasia and < 5 % blasts. If no dysplasia, MDS- associated karyotype |
Table 25.6
Significant changes in the diagnosis and classification of MDS/MPN
1.Some cases of CMML with eosinophilia are relocated to the category “Myeloid/lymphoid Neoplasm with eosinophilia and PDGFRA,PDGFRB. FDGF1 rearrangement” |
2.The category atypical CML has been renamed atypical CML, BCR-ABL 1 -negative to emphasize that it is not merely a variant of CML, BCR-ABL 1-positive |
3.RARS-T remains as a “provisional entity” classified as MDS/MPN, unclassifiable, RARS-T. The criteria have been modified to include refractory anemia, dyserythropoiesis in the bone marrow with ring sideroblasts accounting for 15 % or more of erythroid precursors, and megakaryocytes with features resembling those in PMF or ET: the platelet threshold is lowered to 450,000 cmm |
Table 25.7
Myelodysplastic/myeloproliferative neoplasms (MDS/MPN)
Chronic myelomonocytic leukemia (CMML) |
Diagnostic criteria of AMML |
1.Persistent PB monocytosis > 1 × 109/l |
2.No Philadelphia Chromosome or BCR-ABL 1 fusion gene |
3.No rearrangement of PDGFRA, PGDFRB, or FGFR1 should be specifically excluded in cases with eosinophilia |
4.Less than 20 % blasts in PB and in BM. Blasts include myeloblasts, monoblasts, and promonocytes |
5.Dysplasia in one or more myeloid lineages. If dysplasia is absent the diagnosis of CMML may be made if the other requirements are met and: a.An acquired, clonal cytogenetic or molecular genetic abnormality is present b.The monocytosis has persisted for at least 3 months c.Other causes of monocytosis have been excluded |
Patients with MDS/MPN usually present with a leukocytosis and hepatosplenomegaly. The MDS/MPN category includes four defined entities: chronic myelomonocytic leukemia (CMML), atypical chronic myelogenous leukemia (aCML-Philadelphia chromosome negative), juvenile myelomonocytic leukemia (JMML), and a more heterogenous group of unclassifiable MDS/MPN (U-MDS/MPN) [82–86]. The WHO recently proposed the provisional entity refractory anemia with ring sideroblasts (ringed was changed to ring) and marked thrombocytosis (RARS-T) [87]. The JAK 2 mutation has been noted in some of the cases of CMML, RARS-T, and aCML but the proliferative potential of most cases appear to be related to aberration in the RAS/MAPK or other signaling pathways and the JAK 2 mutation is not the primary oncogenic event to explain the MPN [86]. The MDS/MPN disorders have a variable course but are generally associated with poorer prognosis than their myeloproliferative neoplastic counterpart and are associated with an increased incidence of leukemic transformation [84, 85].
CMML is characterized by both proliferative and dysplastic features. The FAB group recognized this subtype as one of the MDSs. The WHO placed this entity in the overlap group of MDS/MPN. CMML is generally characterized by anemia, hepatomegaly, splenomegaly and the frequent finding of tissue infiltration by monocytic cells. The peripheral blood demonstrates a monocytosis (>1 × 109/l). In addition, to distinguish this disorder from a myeloproliferative neoplasm, the Philadelphia (Ph) chromosome is absent and there is no gene rearrangement of PDGFA or PDGFB. The bone marrow and PB has <20 % blast cells, Monoblasts plus promonocytes and dysplasia is present in one or more myeloid lineage. Cases of CMML with esosinophila and the PDGBFRA, or PDGFRB rearrangement have been removed from the MDS/MPN category and placed in a separate genetically defined group of disorders associated with a rearrangement of PDGFB at 5q31–33. The clinical features of these MPNs are similar to CMML or aCML but are usually associated with a t(5;12) with formation of an ETV6-PDGFRB fusion gene [84, 85]. The cases of CMML that do not demonstrate dysplasia must have a clonal cytogenetic or molecular genetic abnormality in the BM or present with a monocytosis of at least 3-month duration and other causes of monocytosis have been excluded. In many patients the diagnosis of CMML is made by excluding other MPN or reactive causes of a monocytosis. Many patients will present with prominent splenomegaly and an elevated total white blood count. The monocytes are often morphologically abnormal with hypersegmented and atypical nuclei. The monocytes may also appear immature with increased cytoplasmic basophilia and prominent cytoplasmic granules; promonocytes may be noted in the blood and bone marrow but monoblasts are rare. The bone marrow is hypercellular with increased and prominent promonocytes but mature monocytes may not be increased. The atypical monocyte precursors can be difficult to distinguish from dysplastic promyelocytes and mature monocytes may be rare in the BM. In these instances, esterase cytochemistry may be helpful in differentiating promonocytes, esterase positive, from promyelocytes, esterase negative cells. Immunohistochemisty staining for CD 34 is of little value since promonocytes are usually CD 34 negative. Reticulin fibrosis is often diffusely increased on the bone marrow biopsy. Immunological abnormalities are common in this subtype with about half the patients demonstrating autoantibodies, monoclonal proteins, hypergammaglobulinemia, and an elevated erythrocyte sedimentation rate [82]. Patients may present with fevers, pleural or pericardial effusions, ascites, or synovitis. There may be nodules of plasmacytoid dendritic cells in the BM which are part of the neoplastic clone and may reflect the aberrant immune reactivity in CMML. Lymphadenopathy and skin and gum infiltration with abnormal monocytes may be a presently finding. Fatigue, weight loss, fever, and night sweats are common presenting symptoms and patients are frequently misdiagnosed with either a chronic infection or a rheumatologic disorder. Hepatomegaly and splenomegaly are more common in this subtype than the other types of MDS. The WHO recently subdivided the CMML into CMML-1 and CMML-2 based on the percent of blasts plus promonocytes in the PB and BM: CMML 1 = <5 % blasts + promonocytes and < 10 % blasts + promonocytes in the PB and BM respectively: CMML 2 = >5 blasts + promoncytes and > 10 % blast + promonocytes in the PB and BM respectively. The overall median survival is 12–24 months but the prognosis and survival are variable reflecting the clinical heterogeneity of CMML [88]. The administration of steroids may be helpful in treating many of the symptoms and inflammatory changes associated with CMML.
Juvenile myelomonocytic leukemia (JMML) generally presents in children between 3 months and 6 years of age with a median age of 2 years. Children present with hepatosplenomegaly, cough, lymphadenopathy, fever, and diarrhea. Infiltration by normal appearing monocytes into nonhematopoietic organs is common [89]. The diagnosis requires that the PB monocyte count is greater than 1,000 cmm, the Philadelphia chromosome and BCR-ABL gene rearrangement are negative, and the BM has less than 20 % blasts. In JMML, a majority of the patients have mutations of PTPN11, NRAS, KRAS, or NF1 all which encode signaling proteins in RAS-dependent pathways [90]. In majority of cases, the white blood count is elevated with dysplastic monocytes without a basophilia and anemia and thrombocytopenia are common. Monosomy 7, deletion of 7q, and other chromosome 7 abnormalities occur in approximately 25–30 % of cases. Children with neurofibromatosis type 1 have a 500-fold increased incidence of JMML or related myeloid disorders [89]. Genetic lesions in Ras signaling and aberrant Ras signaling pathways are associated with the hypersensitivity of myeloid progenitors to GM-CSF in JMML [89]. The clinical course is heterogeneous but a majority of the children will progress to AML within 3 years of diagnosis and therefore patients should be considered for an allogeneic stem cell transplant early in their clinical course.
Atypical CML (aCML) is a BCR–ABL 1, Ph+ chromosome negative rare MDS/MPN that is characterized by leukocytosis with dysplastic neutrophils. At least 10 % of the leukocytes should be dysplastic. Most of the patients present with anemia and splenomegaly. Monocytosis is more marked in aCML than in Philadelphia chromosome CML (Ph + CML). Eosinophilia and basophilia are usually absent but when present (<10 %) do not exclude the diagnosis of aCML. In aCML, the sum of the promyelocytes, myelocytes, and metamyelocytes is >15 % and monocytes are <10 % in the PB. The BM is hypercellular for the patient’s age but is usually less cellular than seen in patients with Ph + CML. Blasts are less than 20 % and the distinction between aCML and CML is not always possible by morphology or clinical finding alone and therefore cytogenetics and molecular genetic analysis are necessary to confirm the absence of the Philadelphia chromosome, BCR–ABL 1 fusion gene or PDGFRA or PDGFRB rearrangement [88].
The diagnosis of myelodyplastic/myelproliferative neoplasm unclassifiable (MDS/MPN-U) includes patients with features of both myelodysplasia and myeloproliferation who cannot be assigned to a more specific category. Patients must meet the criteria of one of the categories of MDS and demonstrate prominent myeloproliferative features without a prior MPN or MDS. The WHO emphasizes that the diagnosis of MDS/MPN-U should not be made in patients who have recently recovered from cytotoxic chemotherapy or have received recent growth factor therapy. A follow-up evaluation in these patients is essential to demonstrate that the changes in the PB and BM are independent of recent treatments or recovery from chemotherapy. The features that distinguish this subgroup include an elevated platelet count or WBC without evidence of BCR–ABL1 or rearrangement of PDGFRA, PDGFRB or FGFR1, del 5q, t (3; 3), or inv3. In most patients, the karyotype and no abnormal mutational studies are noted but the JAK2 V617F mutation may be present in a minority of cases [88].
The WHO provisional entity RARS—T has features of an MPN. The JAK2V617F mutation is detected in over half of the patients with a platelet count of >600,000 cmm [87, 88]. In addition to the JAK 2 mutation some patients demonstrate the MPL mutation and both mutations appear to correlate with the elevated platelet count. In addition, cases with an isolated del (5q) inv (3)(q21q26.2) and t(3:3)(q21;q26.2) are excluded from this diagnosis. The diagnosis of RARS-T requires refractory anemia with thrombocytosis (>450,000 cmm), dyserythropoiesis includes ring sideroblasts which are present in >15 % of the erythroid precursors. Megakaryocytes have features of a chronic myeloproliferative neoplasm and are large, hyperlobulated and atypical. The megakaryocytes are increased in number with large or giant forms with lobated staghorn like nuclei and occur in lose clusters or found adjacent to the endosteum or within sinusoids. The mechanism of anemia is RARS-T is unclear considering that the JAK 2 mutation is frequently associated with erythrocytosis. The diagnosis of RARS-T remains controversial but the disorder has features of a MDS/MPN. RARS-T is a heterogeneous entity and may include a number of disorders including prefibrotic primary myelofibrosis and MDS [87]. The prognosis of patients with RARS-T is probably similar to patients with RARS and better than most of the MDS/MPN disorders. The transformation to AML appears to be uncommon and unlike the MPN there does not appear to be an increase risk of thrombotic complications associated with the elevated platelet count. The treatment options for this subtype are not clear and additional studies are needed to further define the clinical course.
MDS with Fibrosis and Hypocellular MDS
The WHO classification committee did not define separate categories for MDS with marrow fibrosis or marrow hypocellularity, because of the lack of consensus on the precise definition or the importance of these findings as distinct entities. The majority of patients with MDS present with a hypercellular or normal cellular BM for the patient’s age with ineffective hematopoiesis. A subset of patients present with a hypocellular BM (less than 15 % cellularity on bone marrow biopsy) and minimal dysplasia. Hypocellular MDS must be differentiated from aplastic anemia and hypocellular AML [91–93]. Hypocellular MDS and aplastic anemia may also be pathophysiologically related [93, 94]. The similarity of both disorders is suggested by their response to immunosuppressive therapy. Hypocellular MDS may represent an intermediate stage in the evolution of aplastic anemia. However, MDS tends to occur in older patients, with a more gradual onset and dysplasia involves more than one cell line. Dysplastic megakaryocytes are also more prominent in MDS than in aplastic anemia [95]. Megaloblastic red cell precursors, pancytopenia, and the presence of paroxysmal nocturnal hemoglobinuria (PNH)—like cells occur in both hypoplastic MDS and aplastic anemia and are therefore not helpful diagnostic features [96]. A characteristic MDS cytogenetic abnormality sometimes helps to define MDS but similar findings may also be present in aplastic anemia [97]. There is a link to the DR15 HLA haplotype and the PNH clone and hypocellular MDS. Patients who are HLA-DR15 positive or have a PNH clone respond better to immunosuppression [96]. Patients who do respond to immunosuppressive therapy remain at risk for the late development of other disorders including AML and PNH reflecting the abnormal clonal disorder that remains after treatment [96].
MDS with fibrosis (MDS-F) can be difficult to differentiate from primary myelofibrosis (PMF), acute megakaryocytic leukemia or acute panmyelosis [98–101]. The bone marrow is usually not aspiratable (dry tap) and the morphological findings of dysplasia may be difficult to identify on the BM biopsy. Splenomegaly and leukoerythroblastosis on peripheral smear are unusual in MDS-F and when present suggest the diagnosis of PMF. The abnormal megakaryoblasts can be identified by staining the marrow biopsy and circulating blast forms with megakaryocytic lineage- specific antigens such as factor VIII or GP IIb/IIIa [99]. The finding of diffuse fibrosis in MDS is associated with a poor prognosis and greater than 2+ reticulum fibrosis, as defined by the European consensus system, is an independent negative prognostic marker in MDS [100]. The diagnostic challenges that such cases present are discussed by the WHO with a recommendation that hematopathologists should specifically comment upon hypocellularity or extensive fibrosis in interpretative reports. The WHO classification, however, does not recognize hypocellular MDS and MDS-F as distinct entities as the sub-classification of these cases can be problematic and there is no accepted agreement on the diagnostic features associated with these two disorders. Immunohistochemical stains for CD 34 on the biopsy may demonstrate excess blasts and may help in identifying these patients. BM fibrosis is associated with a higher transfusion requirement, multilineage dysplasia, pancytopenia, and a poor prognosis in most treatment trials [100]. These issues, about when to call a disorder a separate diagnostic entity, raises the question of when a particular disease feature is distinctive enough to warrant a separate diagnostic category or dictate therapy.
The recognition and enumeration of blast cells is of critical importance for the diagnosis of AML, MDS and defining the subtypes of MDS. The WHO classification requires differential blast count of 500 cells on the aspirate to be 20 % or more for a diagnosis of AML (either de-novo or evolved from a prior MDS). If a concomitant non-myeloid neoplasm (i.e., plasma cells) is present those cells should be excluded from the count used to evaluate the percent of blast forms [102]. If an aspirate is not available a touch preparation of the biopsy may yield valuable cytologic information, but differential counts from touch preparations may not be representative and should be confirmed on the BM biopsy. The definition of a blast cell can be difficult in MDS. The FAB group initially defined type I, and II blast cells [101, 102]. Type I and II blasts have a high cytoplasmic nuclear ratio, diffuse chromatin pattern and visible nucleoli without identifiable Golgi formation; Type I blasts are agranular and type II have scanty primary granules. The WHO did not specifically define the definition of a blast, but noted that blasts can be granular or agranular [101]. Myeloblasts are defined by a high nuclear/cytoplasmic ratio, visible nucleoli and usually fine nuclear chromatin. Nuclear shape can be variable and basophilic granules or Auer rods by be present but no Golgi zone is detected. Granular blast cells must be distinguished from promyelocytes and the principal distinguishing characteristic of the normal promyelocyte is the presence of a visible Golgi zone [103]. Dysplastic promyelocytes have the recognizable features of promyelocytes including round, oval or indented nucleus that is often eccentric, it may have reduced granules or irregular distributed granules and a poorly developed Golgi zone. Determining the overall percentage of blasts in the context of marked erythroid hyperplasia can be problematic. When erythroid precursors comprise 50 % of cells in bone marrow then the percent of blasts is based on the non-erythroid cells. If blasts comprise >20 % of the non-erythroid cells, then the WHO criteria for AML (erythroleukemia) is satisfied [103, 104]. As an illustration, 10 % blasts counted from all cells, would correspond to 50 % blasts from non-erythroid cells when erythroid component represents 80 % of all bone marrow cells allowing for a diagnosis of erythroleukemia. The expansion of erythroid compartment can occur in non-neoplastic disorders, including hemolytic anemia, iron, B12 or folate deficiency. Erythropoietin administration may also lead to a marked expansion of the erythroid compartment and blast forms can increase following growth factor administration [104–106]. Simultaneous or chronologically close administration of erythropoietin and granulocyte growth factors may lead to erythroid hyperplasia with an increase in pronormoblasts and myeloblasts [106, 107]. Therefore, the pathologist must have knowledge of the patients clinical history, including the administration of recent chemotherapy or growth factors when evaluating the bone marrow.
The WHO recognized the distinct clinically and pathological differences in MDS in children. In contrast to adults, isolated refractory anemia is uncommon in children. Thrombocytopenia and/or neutropenia with a hypocellular BM are more common in children [108]. The WHO includes the provisional diagnosis of refractory cytopenia of childhood (RCC). This entity includes children with cytopenias and less than 2 % blasts in the PB and less than 5 % in the BM and dysplasia in two or more lineages. The BM is usually hypocellular and in the absence of MDS related cytogenetic abnormalities the distinction between aplastic anemia, congenital marrow failures syndromes and RCC can be difficult. The Recently revised WHO classification defined and redefined a number of new subgroups [109, 110]. Three categories of refractory cytopenia with unilineage dysplsia (RCUD) are defined: Refractory anemia (RA), refractory neutropenia (RN) and refractory thrombocytopenia (RT). These subgroups do not have an increase in blasts and involve a single lineage. Other causes of dysplasia need to be addressed and excluded before the diagnosis of MDS is established (Table 25.8). These include drugs, toxin exposures, viral infections, growth factors, immunologic and congenital disorders, vitamin and trace element deficiencies [111–114]. Identification of a specific clonal chromosomal abnormalities help in confirming the diagnosis MDS. Other causes of erythroid dyspoiesis include alcohol abuse (vacuolated erythroid precursors and ring sideroblasts), anti-tuberculosis medications and chloramphenicol. Isolated unilineage dyspoiesis should raise suspicion for secondary causes, including disorders not associated with prescription medications as seen in zinc over consumption leading to copper deficiency. Zinc competes with copper for its carrier, ceruloplasmin, or in primary copper deficiency leading to anemia and neutropenia [115–118]. Copper deficiency can present with pancytopenia with marked vacuolation of erythroid and myeloid precursors, magaloblastoid changes in red cell precursors and ring sideroblasts. Chromosome abnormalities are helpful in differentiating a reactive cause of dysplasia or cytopenias and MDS [119] (Table 25.9). The WHO has defined the widely accepted recurrent chromosomal abnormalities considered as presumptive evidence of MDS in the setting of persistent cytopenias [120]. Of note common cytogenetic abnormalities including del(20q), +8 and -Y are not included because these abnormalities have been reported in other disorders with no clinical or morphologic evidence of MDS even with prolonged follow up [120, 121]. Loss of the Y chromosome in hematopoietic cells may be associated with aging and not necessarily indicative of MDS in the absence of defined morphologic features. Moreover, clonal cytogenetic aberrations can be induced transiently. Spontaneously reversible of refractory anemia with monosomy 7 and marked dyspoiesis in association with azathioprine administration has been described [122, 123].
Table 25.8
Differential diagnosis of myelodysplastic syndromes (MDS)
Disorder | Comments |
|---|---|
Congenital dyserythropoietic anemia | Inherited disorder characterized by marked dyspoiesis of mature and immature erythroid elements. Granulocytes and megakaryocytes unremarkable. Unstable hemoglobins Normal karyotype. |
Aplastic anemia | Both congenital (Fanconi) and acquired aplastic must be distinguished from hypocellular myelodysplasia. Karyotype normal in acquired aplastic anemia, rare cases with clonal abnormalities may actually represent hypocellular myelodysplasia. Severe malnutrition. |
Hypocellular neoplasms | Hypocellular MDS must be distinguished from hypocellular AML. MDS with fibrosis may resemble primary myelofibrosis. |
Megaloblastic anemia | Dyspoiesis restricted to megaloblastic changes. Normal karyotype. Use of antimetabolites need to confirm normal B12 level. Antimetabolites, Chemotherapy. |
Toxic exposure | Arsenic poisoning, Alcohol, Chemotherapy Bone marrow cellularity normal; blasts not increased. Normal karyotype; normal vitamin B12 and folate levels. Neurologic and gastrointestinal manifestations may Dominate clinical picture. |
Disorders with ringed sideroblasts | Chemotherapy, Copper deficiency, Hereditary sideroblastic anemia. Pyridoxine deficiency, zinc toxicity, alcohol toxicity, and in patients receiving Antituberculosis agents, chloramphenicol. Heavy metals. |
Fibrotic disorders | Acute megakaryoblastic leukemia. Metastatic carcinoma with marked fibrosis, Hairy cell leukemia, Primary myelofibrosis. |
Viruses | Single, double or trilineage dyspoiesis common. HIV-1. Parvovirus. Karyotype normal |
Autoimmune disorders | Myelodysplasia-like picture with dyspoiesis may be found in patients with underlying immune defects. May precede evolution to red cell aplasia. SLE, rheumatoid arthritis. |
Paraneoplastic syndromes | Myelodysplasia-like picture (usually resembling chronic myelomonocytic leukemia). Occasionally noted at diagnosis in patients with solid tumors (lung, colon, prostate, and gastric carcinoma and lymphomas). Distinct from therapy-induced bone marrow neoplasms in carcinoma patients. |
Bone marrow regeneration | Abnormal localization of immature precursors, dyspoiesis and increased immature myeloid cells may be transient phenomena after aggressive therapy. Increased blasts from recovering bone marrow after cytoxic chemotherapy, drug toxicity or bone marrow transplantation. Karyotype normal. |
Colony-stimulation factor therapy | Transient increase in blasts in blood and bone marrow. clusters of blasts on core biopsy. Hyperlobated neutrophils and toxic granule in neutrophils may be seen. |
Other drug treatments | After purine analog therapy, Zidovudine and other antivirals Dilantin, methrotrexate, valproic acide, sulfasalazine, mycophenolyte, etc. |
Other | HIV infection, Tuberculosis, multiorgan failure, post chemotherapy effects, Radiation to bone marrow, PNH, Storage diseases. Acute panmyelosis with myelofibrosis. |
Table 25.9
Recurring chromosomal abnormalities considered presumptive evidence of MDS in the absence of definitive morphological features
Abnormality | WHO-estimated frequency in MDS (%) |
|---|---|
Unbalanced | |
−7 or del(7q) | 10 |
−5 or del(5q) | 10 |
i(17q)or t(17p) | 3–5 |
−13 or del(13q) | 3 |
del(11q) | 3 |
del(12q) or t(12q) | 3 |
del(9q) | 1–2 |
idic(x)(q13) | 1–2 |
Balanced | |
t(11;16) | 3 in t-MDS |
(q23;p13.3) | |
t(3;21) | 2 in t-MDS |
(q26.2;q22.1) | |
t(1;3) | <1 |
(p36.3;q21.2) | |
t(2;11)(p21;q23) | <1 |
inv(3) | |
(q21q26.2) | <1 |
t(6;9)(p23;q34) | <1 |
In patients with RCUD without clonal cytogenetic abnormalities or atypical presentations the WHO recommends a period of observation of 6 months before the diagnosis of MDS is established. The presence of PB blasts excludes the diagnosis of RCUD. However, an occasional patient may present with 1 % blast forms in the PB and <5 % blasts in the BM. If on two successive evaluations blasts are noted then the patient is more appropriately classified as MDS—unclassified (MDS-U). The diagnosis of MDS-U reflects the uncertainty of the clinical course and biology of these findings. Patients with one unilineage dysplastic subtypes may demonstrate bi-cytopenia in the PB but with morphologic dysplasia of a single lineage. However, the classified as MDS-U is more appropriate if the patient is pancytopenic even in the context of unilineage dysplasia. The MDS-U subtype includes disorders with unequivocal dysplasia in <10 % of cells in one or more lineage with accompanied cytogeneic abnormality.
Idiopathic Cytopenia(s) of Undetermined Significance and Idiopathic Dysplasia of Uncertain Significance
Patients who present with cytopenias but lack the diagnostic criteria of MDS, but in whom the diagnosis of MDS is suspected, are diagnosed with “Idiopathic Cytopenia(s) of Undetermined Significance” (ICUS) (Table 25.10) [124, 125]. The criteria for this group of disorders reflected the diagnostic uncertainty in these patients: A key distinction of ICUS from other potential precursor conditions such as monoclonal gammopathy of undetermined significance (MGUS), monoclonal B-cell lymphocytosis (MBL) and T cell clonality of undetermined significance is that an ICUS designation does not necessarily imply a clonal disorder.
Table 25.10
Summary of a proposed definition of idiopathic cytopenia(s) of undetermined significance
ICUS |
|---|
1.Meaningful cytopenias (hemoglobin < 11 g/dl, absolute neutrophil count) <1,500/mm3, or platelets < 100 × 109/l Lasting more than 6 months |
2.Does not meet minimal diagnostic criteria for MDS |
3.Other recognized causes of cytopenias have been excluded (see listing that follows) |
“Required” investigations to exclude other causes of cytopenias |
1.Detailed case history (toxins, drugs, mutagenic, events, etc.) |
2.Thorough clinical investigations, including X-ray or sonography of spleen |
3.Differential blood count (microscopic) and complete serum chemistry |
4.Bone marrow smear, histology and immunohistochemistry |
5.Flow cytometry of bone marrow and peripheral blood cells |
6.Chromosome analysis including FISH |
7.Molecular analysis where appropriate (for example, T-cell receptor rearrangement for neutropenia) |
8.Exclusion of viral infections (HCV, HIV, CMV, EBV, others) |
Limited data are available about the frequency or natural history of ICUS and reflect that some patients present with persistent cytopenia but lack the diagnostic features of MDS.
The term idiopathic dysplasia of uncertain significance (IDUS) was proposed to describe a group of patients with dysplasia but no or only mild cytopenias [125]. In contrast to ICUS, patients with IDUS demonstrate dysplasia in >10 % of cells in one or more lineage with or without a MDS-related karyotype but without persistent cytopenias. ICUS and IDUS have a very variable course and it is unclear if all the patients will ultimately develop MDS or another myeloid neoplastic disorder. Moreover, it is unclear if these disorders as mutually exclusive or if the classification of potentially premalignant disorders will provide meaningful prognostic or diagnostic information for patients in whom no cause for the cytopenia is found [111, 125].
MDS Subtypes as Defined by the WHO
Refractory Anemia with Ring Sideroblasts
Refractory anemia with ring sideroblasts (RARS) is characterized by anemia, erythroid dysplasia and > 15 % ring sideroblasts of bone marrow erythroid precursors. There is no dysplasia in the non-erythroid precursors. Myeloblasts comprise < 5 % of the nucleated BM cells and are not present in the PB. RARS constitutes approximately 10 % of cases of MDS. A majority of patients present with a moderated normochromic or macrocytic anemia. The PB frequently reveals dimorphic red cells due to a small population of microcytic and hypochromic red cells. Basophilic stippling and Pappenheimer bodies may be noted in red cells. Dysplasia is present in <10 % of neutrophils and platelets. The bone marrow is usually hypercellular for the patient’s age and demonstrates erythroid hyperplasia. The iron stain, Prussian blue staining, reveals that >15 % of erythroblasts are ring sideroblasts and contain >5 iron containing granules surrounding at least a third of the nuclear circumference. Iron stores are generally increased. The number of CD 34 positive cells is normal and most patients do not demonstrate a cytogenetic abnormality. Ring sideroblasts may be seen in a number of other, non- MDS-related disorders. Lead poisoning, drugs including isoniazid inhibit delta aminolevulinic acid (ALA) dehydratase activity and block hemoglobin formation resulting in ring sideroblast formation [126]. A number of acquired and hereditary conditions are associated with ring sideroblast formation and should be excluded before a diagnosis of RARS is established. In RARS the ring sideroblasts and increased iron stores reflect abnormal iron metabolism in the erythroid lineage resulting from the ineffective erythropioesis. The overall prognosis for patients with RARS is 69–108 months and less than 2 % of cases transform into AML. Progressive anemia requiring transfusion support is frequent and in select patients iron chelation therapy should be considered early in the clinical course to prevent iron overload and end organ failure [127]. However, the overall beneficial effects of early iron chelation remain controversial [126, 127].
Refractory Cytopenia with Multilineage Dysplasia
Refractory cytopenia with multilineage dysplasia (RCMD) constitutes approximately 30 % of MDS cases and is characterized by one or more cytopenias and dysplastic changes in two or more of the myeloid lineages. Blasts are rare, <1 %, in the PB and <5 % in the BM. Auer rods are not present in either the PB or BM. The anemia is usually macrocytic or normocytic with prominent granulocytic dysplasia including hypogranularity, nuclear shape abnormalities including hypolobation, acquired pseudo pelger-huet anomaly, and abnormal nuclear clumping. The bone marrow is usually hypercellular for age of the patient with < 5 % blasts. Erythroid precursors may demonstrate cytoplasmic vacuoles and marked nuclear irregularity including internuclear bridging and nuclear budding. The BM may have variable number of ring sideroblasts. The previously described WHO category of refractory cytopenias with multilineage dysplasia with ring sideroblasts has been omitted and incorporated in RCMD. The presence of > 15 % ring sideroblasts did not change the prognosis or clinical course in patients with RCMD. Megakaryocytic dysplasia including hypolobated and non-lobated nuclei, multinucleated and micromegakaryocytes, megakaryocytes with non-lobated or bi-lobed nuclei. Clonal cytogenetic abnormalities are present in up to 50 % of patients and are important in defining the prognosis. The prognosis is related to the degree of cytopenias and cytogenetic abnormalities [128]. The overall survival in approximately 30 months.
Refractory Anemia with Excess Blasts (REAB 1 and 2)
RAEB comprises 40 % of cases of MDS and is divided into RAEB-I and RAEB-2 on the basis of the number of blasts and the presence or absence of Auer rods. RAEB 1 is defined by 5–9 % blasts in the BM or 2–4 % blasts in the PB and no Auer rods. RAEB -2 is defined by 10–19 % blasts in the BM or 5–19 % blasts in the PB. The presence of Auer rods confirms the diagnosis of RAEB 2 irrespective of the percent of blast forms. Most patients present with symptoms of BM failure including anemia, bleeding or neutropenia. The PB generally shows dysplastic changes in all three cell lines and is typically hypercellular for age of the patient. Erythroid precursors may be increased with megaloblastoid changes and ring sideroblasts. The excess blasts define these subtypes. Dysmegakaryopoiesis is a frequent finding including micromegakaryocytes and abnormal megakaryocytic clustering. Blasts may form abnormal aggregates or clusters that are located away from trabeculare and vascular structures, a histologic finding previously referred to an abnormal localization of immature precursors (ALIP). Immunohistochemical staining for CD 34 have help in identifying blast forms. Fibrosis may be present and result in a dry tap. MDS with fibrosis is diagnosed when there is accompying coarse reticulum fibrosis with or without collagen fibrosis. The presence of fibrosis should be noted and the finding of extensive fibrosis is an independent negative prognostic marker in MDS [100].
Distinct Subtypes Recognized by the WHO
Myelodyplastic Syndrome with Isolated del (5q)
MDS with isolated del (5q) was formerly called the 5q- syndrome, and is a favorable prognostic subtype of MDS defined by the specific cytogenetic abnormalities, the loss of all or part of the long arm of chromosome 5 [129, 130]. The subtype MDS with isolated del (5q) is a distinct clonal disorder. In contrast, patients with del (5q) and additional chromosomal abnormalities and excess blast have a different clinical course and response to treatment [131]. The long arm, q arm, of chromosome 5 contains the genes that encode for a number of growth factors including interleukin (IL)-3, IL-5, IL-9, and granulocyte-macrophage colony-stimulating factor (GM-CSF) [130]. The 5q—deletion occurs at the level of the multipotent stem cell and is associated with a deletion at 5q31–32. The deleted region results in haplo-inusfficiency for the ribosomal gene RPS 14 resulting in a specific block in erythroid maturation. RPS 14 is a component of the ribosomal 40s subunit and down regulation of RPS14 leads to defective erythropoiesis and increased apoptosis in erythroid progenitors. Moreover, in the congenital disorder Diamond- Blackfan anemia the down regulation of an another ribosomal gene (RPS19) is critical in the development of the erythroid hypoplasia and chronic anemia [132]. The down regulation of RPS14 may not be the sole genetic event underlying the del 5q- syndrome and alteration of other genes in the commonly deleted segment in 5q- may be required. The tumor suppressor SPARC (Secreted Protein Acidic and Rich in Cysteine) gene is located in the del 5q31 region. SPARC has tumor suppressor, antiproliferative, and anti angiogenesis properties and may also be important in this syndrome [130]. The loss of additional genes that code for these and other factor appear to contribute to the development of this syndrome and its unique response to the immune modulatory drug, lenalidomide. MDS with isolated del 5q syndrome is frequently associated with morphological features of RA and to a lesser extent RARS and the other subtypes. Thrombocytosis and anemia is occasionally seen and when present is suggestive of the del 5q chromosomal abnormality. The bone marrow aspirate and biopsy are typically hypercellular for the patient’s age with dysplastic erythropoiesis. Bone marrow blasts constitute less than 5 % of all cells and less than 1 % of blast forms are in the blood. Ring sideroblasts may be present but do not exceed 15 % of erythroblasts. A majority of the patients present with a refractory macrocytic anemia, with a normal or mildly low leukocyte counts, thrombocytosis (a platelet count >400,000 cmm), and less than 5 % blast forms in the bone marrow. In contrast to the other MDS subtypes, where the mononuclear megakaryocytes are smaller (micromegakaryocytes), in the MDS with del 5q the megakaryocytes are bilobed or non-lobulated but of normal size (monolobulated). The MDS with del 5q has a marked female predominance (70 %), and rarely transforms to AML. A majority of patients have progressive anemia and become red cell transfusion dependent. Particular attention should be paid to managing and preventing iron overload early in the course of the disease which otherwise has a favorable prognosis. Lenalidomide is the treatment of choice and results in transfusion independence in over two thirds of cases [129].
Therapy-Related Myeloid Neoplasms
MDS that occurs following exposure to cytotoxic chemotherapy and/or ionizing radiation is referred to as secondary-therapy-related MDS (t-MDS). Secondary MDS should be considered as entity separate from primary-de novo MDS. The WHO has assigned t-MDS to the major subgroup of acute myeloid leukemia. The therapy related myeloid neoplasms are separate from acute myeloid leukemia with myelodyplastic related changes. Patients with AML who have evolved from a previously documented MDS are assigned to this later category. The WHO considers the therapy-related myeloid neoplasms as a unique clinicopathologic syndrome—therapy-related MDS/AML. A variety of cytotoxic agents are implicated in the development of t-MDS/AML. The standard MDS classification, prognostic indicators, and approaches to treatment are not applicable in patients with t-MDS [73, 133]. MDS secondary to alkylating agents is associated with deletions or loss of chromosomes 5, 7, and 8, has a latency period of 3–7 years, and usually progresses to AML. In contrast the topoisomerase II inhibitors, including the epipodophyllotoxins and anthracyclines, are associated with balanced translocations involving chromosome bands 3q26, 11q23, and 21q22 and a latency period of 18 months to 3 years after exposure [134]. The clinical presentation of these two types of therapy-related disorders is also different. The alkylator-related disorders present initially with a slowly progressive illness with many features of de novo MDS including trilineage dysplasia and pancytopenia. In contrast, the topoisomerase II associated disorders rarely present with features of de novo MDS and rapidly evolves to AML. The prognosis of all t-MDS is very poor. Patients do not respond to the standard treatments for MDS and an allogeneic transplant, if a donor is available, should be considered as the primary therapy for all eligible patients.
Clinical and Laboratory Features of MDS
A majority of patients with MDS are asymptomatic at presentation and are usually diagnosed when they present with an unexplained macrocytic anemia (MCV >102) and an absolute low reticulocyte count. Anemia is the most common presenting abnormality and patients may complain of insidious onset fatigue and progressive dyspnea on exertion. Bone pain and weight loss are uncommon. While patients may be neutropenic and have dysplastic and impaired neutrophil function, infections are unusual at presentation. The physical examination is notable for the lack of adenopathy, cutaneous lesions, prominent splenomegaly, or hepatomegaly. The diagnosis of MDS relies largely on the morphological findings in the peripheral blood and the bone marrow. MDS must be differentiated from other disorders that present with abnormalities of one or more cell line including aplastic anemia, myeloproliferative disorders, nutritional deficiencies, and autoimmune disorders including system lupus erythematosus (Table 25.8). AML in elderly patients may also present with progressive pancytopenia with rare circulating blast forms [135]. The differentiation of AML with dysplasia from MDS can be difficult. Patients with acute erythroleukemia may have prominent dysplastic erythroid precursors and may initially be diagnosed with one of the MDSs. Patients with hypoplastic AML can also be confused with one of the MDS subtypes. In prospective trials of MDS up to 15 % of the patients with hypoplastic MDS were later reclassified as having AML [136].
The finding of a clonal cytogenetic abnormality characteristic of MDS is important in establishing the diagnosis, assessing prognosis and differentiating it from other disorders [137]. The progress in fluorescence in situ hybridization (FISH) techniques has facilitated cytogenetic diagnostics and compliments karyotyping. FISH can be performed on the non dividing cells and therefore can be performed on PB cells. The study of interphase cells may be helpful in identifying specific rearrangements not recognized by banding studies alone. FISH provides increased sensitivity for monitoring patients. A major disadvantage of FISH studies is that the analysis is restricted to specific probes, defined chromosomal abnormalities. The patient’s clinical findings and history are helpful in guiding the use of FISH analysis; in patients with a macrocytic anemia and thrombocytosis the possibility of del(5q) should be suspected and del(20q) is associated with thrombocytopenia and dysplastic erythroid cells [138]. The diagnosis of MDS requires the collaborative efforts of clinicians, pathologists, and cytogeneticists. Cytogenetics analysis, standard karyotype and FISH analysis should be a routine part of the initial evaluation of all patients with MDS. Detectable clonal cytogenetic abnormalities are found in 38–78 % of de novo MDS and greater than 80 % of t-MDS patients [119]. Cytogenetics are also helpful in evaluating disease progression and prognosis. The acquisition of new cytogenetic abnormalities is generally associated with progression of the disease and a poor prognosis. Although anemia is the most common presenting laboratory feature in patients with MDS, approximately 50 % of patients will demonstrate a abnormality of more than one cell line [139]. The white blood count is usually low, but rarely less than 1,000 cmm. Patients may be at risk for bacterial infection due to qualitative abnormalities of neutrophil function. Platelet functional abnormalities are associated with increased risk for bleeding even with an adequate platelet count. Iron studies may demonstrate increased iron stores especially in patients with the refractory anemia with ring sideroblast subtype.
Blood and Bone Marrow Findings
Red Cells/Anemia
Macrocytosis or a macrocytic anemia with a low reticulocyte count are common in MDS and due to ineffective erythropoiesis. Impaired red cell maturation has been associated with acquired abnormalities of globin chain synthesis, and red cell enzymes [140]. PNH has been described in the setting of MDS, and these patients have many of the typical diagnostic features of PNH including a defect in the synthesis of the glycosylphosphatidylinositol (GPI)-linked surface protein, but lack the ongoing red cell hemolysis and thrombotic complications associated with PNH [141]. On the peripheral blood smear patients may have abnormalities in the size and shape of red cells including basophilic stippling (red cell inclusions composed of ribonucleoprotein and mitochondrial remnants), Pappenheimer bodies (basophilic iron-containing granules peripherally located in red cells), macro-ovalocytes, teardrop forms, and nucleated red cells. The bone marrow may reveal ring sideroblasts, multinuclear fragments, inter-nuclear bridging, and nuclear cytoplasm asynchrony.
Neutrophils
Qualitative abnormalities of neutrophil function are a common feature of MDS and may explain the increased risk for bacterial infections [142]. Morphological abnormalities include hypo-granular and hyposegmented neutrophils, which are associated with a negative peroxidase reaction and decreased myeloperoxidase activity. The neutrophils are hyposegmented and may be confused with band forms. Nuclear fragmentation and nuclear-cytoplasmic asynchrony in early myeloid precursors may be a prominent feature in the bone marrow. Dysplastic myeloid precursors can be difficult to distinguish from blast forms and therefore the bone marrow should be reviewed by a pathologist experienced in the interpretation of MDS.
Platelets
Thrombocytopenia and abnormal platelet function occur in MDS. Thrombocytopenia is an adverse prognostic feature independent of other prognostic factors [143]. While thrombocytopenia is associated with poor performance status and other unfavorable prognostic variable bleeding complications are underreported [143]. Thrombocytopenia (<100,000 mm) has been reported in 66 % of patients and was associated with a 24 % incidence of deaths from hemorrhage. Impaired platelet function may also explain the increased risk of bleeding in patients with MDS. Spontaneous bruising and bleeding after surgery or mild trauma occurs in MDS patients with a normal or slightly depressed platelet counts [143, 144]. Dysplastic platelets and abnormal megakaryocytes are important diagnostic features and help in distinguishing MDS from other disorders. Giant platelets, and agranular (grey platelets) and megakaryocytic fragments in the peripheral blood film are important diagnostic features in MDS.
Bone Marrow Findings
A bone marrow aspirate and biopsy is essential for diagnosis and to define the MDS subtype. Cytogenetics should be performed to assign prognosis and differentiate MDS from other disorders. The bone marrow is usually hypercellular but may be normal or hypocellular for the patient’s age. Abnormal distribution of cells is often present; erythroid islands may be absent or very large. Granulocytic precursors may be clustered centrally rather than their normal paratrabecular distribution. This phenomenon has been designated as abnormal localization of immature precursors (ALIP) and is associated with a poorer prognosis [145, 146]. The presence of ALIP can be helpful in distinguishing MDS from other myeloid neoplasms. Micromegakaryoctes, mononuclear megakaryocytes, and hyperlobulated megakaryocytes are important diagnostic features of MDS and are reliable morphological evidence of dysplasia in the bone marrow smear and biopsy. The megakaryocytes may be clustered or adjacent to the bony trabeculum. The del 5q syndrome has mononuclear megakaryocytes that are of normal size but with a single eccentrically placed round nonlobulated nucleus [130]. Megaloblastic changes (nuclear cytoplasm asynchrony) can be seen in the myeloid and erythroid precursors. Dysgranulopoiesis and dyserythropoiesis are more readily noted in the bone marrow aspirate smear and not the biopsy. The bone marrow smear is necessary to identify ring sideroblasts that may not be apparent on the biopsy sample. Immunohistochemistry may be a useful supplement to histology. The use of anti-glycophorin antibodies may be helpful to identify erythroid precursors and differentiate them from ALIP. Antibodies against the von Willebrand factor may recognize small mononuclear megakaryocytes that can be confused with myeloid precursors. A biopsy is necessary to access the degree of reticulin fibrosis and overall bone marrow cellularity. Cytochemistry also can be helpful in some cases, periodic acid-schiff (PAS) stain may reveal positive granules in erythroblasts and diffuse cytoplasmic positivity in more mature erythroid elements. Normal erythroid cells lack any PAS positivity. In cases when immature cells are expanded and their lineage (myeloid, monocytic, erythroid) is difficult to establish, cytochemistry for myeloperoxidase and non-specific esterase on the aspirate, touch imprints and blood smears may be helpful. Myeloid blasts are myeloperoxidase positive, while non-specific esterase positivitiy indicates monocytic differentiation.
Immunophenotyping using flow cytometry on the BM and/or PB may be helpful in the diagnosis and defining prognosis and response to treatment [147, 148]. However, currently there are no accepted standards for the diagnosis of MDS by flow cytometry.
The finding of aberrant immunophenotyping of myeloid blasts is helpful in corroborating the diagnosis of MDS, but is not diagnostic of MDS. The aberrant expression of the lymphoid antigen CD7 on myeloid blasts is a common phenotypic abnormality and correlates with a poor prognosis [126]. Increase and/or clustering of blasts favors MDS. Immunostaining for CD34 on core biopsy is very helpful to estimate blast numbers and possible clustering. In the absence of reliable aspirate smear, CD34 immunostaining on core biopsy and/or clot section can be used for estimating percentage of blasts. Sometimes blasts may be negative for CD34 and identification of immature myeloid precursors can be assisted by CD117 [146]. CD33 and myeloperoxidase can be used to estimate the myeloid/erythroid ratio (in combination with red cell marker), but more mature myeloid cells will be positive for CD33 and myeloperoxidase. Expansion of monocytic cells can be identified by immunostaining panel including CD34, CD33, myeloperoxidase and lysozyme. Monocytic cells are negative for CD34 and weak positive or negative for myeloperoxidase, but positive for CD33 and lysozyme [149]. The use of flow cytometry on both the PB and BM is the focus of many studies and should be part of the initial evaluation of MDS [146–148].
Related posts:


Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


