Chapter 9
The Liver and Pancreas
Jason Beyers
The Liver
Introduction
Liver disease in children differs greatly from that in adults. Klein et al. have summarised the differences [1]:
- liver disease in paediatric patients is rare
- the causes of disease are more diverse
- there is a greater prevalence of inherited metabolic disorders, biliary tract disease, primary infections and autoimmune disorders
- the higher anabolic needs for growth plus high levels of malabsorption and the catabolic effects of liver disease may result in more nutritional deficiencies
The nutritional management of an infant or child will depend on whether the liver disease is acute, chronic or metabolic. Potential problems warranting nutritional attention occur when there is a disturbance in the usual metabolic functions of the liver. These include glucose homeostasis, protein synthesis, bile salt production, lipid metabolism and vitamin storage. Dietary therapy is usually aimed at managing the consequences of these disturbances (Fig. 9.1).
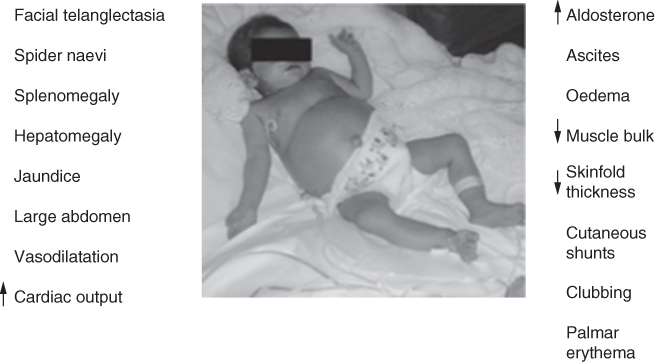
Figure 9.1 Liver disease in children.
Screening and investigations
In the UK, newborn screening is performed in the first week of life using the newborn screening collection card. The number of conditions tested for varies between regions, but includes phenylketonuria, cystic fibrosis and medium chain acyl-CoA dehydrogenase deficiency. Many countries outside the UK use this test for a wider range of conditions.
An infant presenting with conjugated hyperbilirubinaemia (CHB) or a suspected inherited metabolic disorder (IMD) requires a referral to a specialist liver centre. A full range of laboratory screening tests are undertaken to investigate potential liver pathology and up to 150 laboratory tests within the first 24 hours of admission may be initiated (Table 9.1). Additional functional laboratory tests (including biopsy) may also be ordered.
Table 9.1 Core conjugated hyperbilirubinaemia work up tests for a patient less than 6 months old
| Haematology | Biochemistry | Virology | Immunology | Other tests | Functional tests |
| INR | Creatine kinase | Hepatitis markers | Anti tissue transglutaminase | Gal-1-PUT | Liver ultrasound |
| Reticulocyte count | Alpha-1-antitrypsin phenotype | Lymphadenopathy/ Glandular fever screen | Vitamin D | Liver biopsy | |
| Full blood count | Antineutrophil cytoplasmic antibody | Vitamin E | Anteroposterior spine x-ray | ||
| Alpha feto protein | Viral & microbiology serology: CMV IgM, toxoplasma & EBV, HIV (needs consent) | Vitamin A | |||
| Direct antiglobulin test | Bilirubin (conjugated) | Auto antibodies | Skeletal survey | ||
| Group and screen | Lactate (plasma) | Immunoglobulins | Amino acids (urine) | ||
| Lipid profile | Bile acids (urine) | Eye examination (Alagille’s syndrome) | |||
| Biochemistry | |||||
| ALT | |||||
| Renal/Liver/Bone/Urea |
INR international normalised ratio
ALT alanine transaminase
CMV cytomegalovirus
IgM immunoglobulin M
EBV Epstein-Barr virus
HIV human immunodeficiency virus
Gal-1-PUT galactose-1-phosphate uridyl transferase
Biochemistry
Preliminary biochemical tests help identify possible causes of liver disease (Table 9.1) and these test results identify further necessary investigations. Due to the high number of potential causes of liver disease a variety of tests are required to determine the likely diagnosis, e.g. patients presenting with neonatal jaundice will require biochemical tests and a possible ultrasound, whereas a patient who presents with suspected biliary atresia will require more additional invasive procedures including liver biopsy or exploratory laparotomy.
Nutritional assessment
Patients with liver disease require regular nutritional assessment. The frequency of assessment is determined by the stage and severity of liver disease. Acute, acute on chronic and end stage liver disease (ESLD) assessment are highly variable and are discussed below. After liver transplantation progress is monitored closely over the ensuing weeks and constant reassessment is required until full oral feeding is established. By 6 months post liver transplant the majority of children are taking a normal diet.
Children with acute on chronic or advanced ESLD require regular anthropometric measurements for weight, length, head circumference (if < 2 years), mid upper arm circumference (MUAC) and triceps skinfold. Care is needed when assessing weight, which can be affected by organomegaly, oedema and ascites [2]. The plotting of length/height can indicate chronic malnutrition over longer time periods [3] but is often confounded by inter-observer error. Some liver conditions (e.g. Alagille’s syndrome, progressive familial intrahepatic cholestasis (PFIC) and α1-antitrypsin deficiency) predispose the child to short stature, with length often falling well below the 0.4th centile. Serial measurement of head circumference for children < 2 years is an excellent long term measure of nutritional status. MUAC is used as an indicator of muscle and subcutaneous adipose tissue and has the advantage of not being greatly affected by fluid shifts. It is useful as a short term measure of nutritional status [4]; MUAC centiles are available for infants < 5 years of age [5]. Triceps skinfold measurement can be helpful to determine medium to long term nutrition status [6–8]. The use of total body potassium depicts diminishing cell body mass in ESLD patients and is three times more predictive than anthropometry alone [9]. However, it is costly and labour intensive so is not routine practice in most institutions [10].
The severity of malnutrition does not always correlate with measurable biochemical markers such as liver function tests or vitamin and mineral status [11] and many other measures are not useful malnutrition markers in liver disease [12].
Assessment should also include signs of any self-imposed dietary restrictions (particularly for children with Alagille’s or PFIC), early satiety and clinical signs of malnutrition or vitamin deficiencies. It is important to consider any nutrition related problems such as nausea, vomiting, diarrhoea, anorexia or severe itching (e.g. pruritis).
Vitamins and minerals
Fat soluble vitamin deficiencies are a feature of cholestatic and chronic liver disease (CLD). All infants and children with cholestatic liver disease require additional fat soluble vitamins [13] (Table 9.2). Cholestyramine is prescribed to increase gall bladder contraction and this impairs absorption of fat soluble vitamins. Monitoring is essential and additional oral vitamin doses should be given and adjusted accordingly. In severe malabsorption, adequate levels become difficult to achieve and vitamins may need to be given as intramuscular injections.
Table 9.2 Practice guide for vitamin supplementation (King’s College Hospital, London)
| Vitamin | Product | Age | Daily oral dose |
| Multi vitamins (A, B group, C & D) | Abidec/Dalivit drops (given with α-tocopheryl acetate, see below) | Birth-1 year > 1 year | 0.6 mL 1.2 mL |
| Forceval capsule | > 12 years | 1 capsule | |
| Vitamin E | α-tocopheryl acetate (vit E deficiency with cholestasis and severe liver disease) | 1 month-12 years | 10 mg/kg initially (up to 200 mg daily) |
| Vitamin D | Alfacalcidol oral drops 2 µ g/mL (prevention of vit D deficiency in cholestatic liver diseases) | 1 month-12 years (<20 kg) | 15-30 ng/kg (maximum up to 500 ng) |
| or Colecalciferol 3000 IU/mL | 1-6 months 6 months-12 years 12-18 years | 3 000 IU 6 000 IU 10 000 IU | |
| Vitamin K | Menadiol tablets (water soluble Vit K tablet preparation for oral supplementation as liver patients have problems with fat malabsorption) | Infants Children | 1 mg 5-10 mg |
There is limited research regarding water soluble vitamin supplementation. Due to an altered hepatic metabolism, twice the recommended daily allowance for water soluble vitamins has been recommended in CLD [14]. In adequately nourished patients, oral nutritional supplements given to achieve energy requirements will indirectly increase micronutrient intake, possibly alleviating the need to supplement with water soluble vitamins and minerals. However, in cases of faltering growth it would be prudent to monitor levels and supplement as needed. Increased mineral requirements may include iron if bleeding has been a problem; zinc if vitamin A deficiency occurs; and calcium and phosphate if rickets is diagnosed. Selenium deficiency is associated with essential fatty acid (EFA) deficiency.
Acute liver failure
Acute liver failure (ALF) in children was first described in 1996 by Bhaduri and Mieli-Vergani [15] as a heterogeneous and multi-systemic disorder in which severe impairment of liver function, with or without encephalopathy, occurs in association with hepatocellular necrosis in a patient with no recognisable underlying CLD.
Infants tend to present with symptoms of biliary tract disorders that can progress to a chronic condition. Acute presentations are usually due to poisoning, or as a result of an IMD. An acute presentation in an older child may be due to hepatitis infection, ingestion of a toxic substance or to decompensation of an underlying CLD which has been ‘silently’ progressing over time. Examples include Wilson’s disease or autoimmune liver disease.
Causes of ALF
- infective, e.g. hepatitis A, B or C, cytomegalovirus
- IMD including haemochromatosis, galactosaemia, tyrosinaemia
- toxins/drugs, e.g. parenteral nutrition, chemotherapy or paracetamol overdose
- irradiation, e.g. radiotherapy
- ischaemic, e.g. Budd–Chiari syndrome
- infiltrative such as leukaemia
- autoimmune, e.g. autoimmune hepatitis, autoimmune sclerosing cholangitis
- trauma from abuse, a fall from a horse or bicycle, or a seat belt laceration caused by a road traffic accident
All patients presenting with symptoms of ALF are suspected to have an IMD, which are most commonly seen in neonates and young infants. Clinical presentations are varied and require specific dietary treatment. Prevention of hypoglycaemia is the primary aim (see emergency regimen, Table 9.6) as the risk of prolonged or frequent bouts can accelerate irreversible liver or neurological damage. Dietary manipulation of macronutrients is managed according to routine monitoring of blood glucose levels and specific serum laboratory tests until a diagnosis is confirmed. Often, until a diagnosis is made macronutrient provision is advised by the metabolic team, who review blood results daily and advise on upper limits (particularly in disorders of protein metabolism). Once a diagnosis is made lifelong dietary management will be required (Chapters 17–19) unless the child has a condition that can be alleviated by liver transplantation.
Examples of symptoms associated with ALF according to age at presentation and the suspected IMD are listed in Table 9.3.
Table 9.3 Common clinical symptoms at first presentation of acute liver failure in patients with suspected genetic defects
| Age group | Common symptoms | Possible disease according to age of presentation | Age of onset |
| Neonate | Hypoglycaemia, jaundice, poor feeding, vomiting, diarrhoea, lethargy, sepsis, lactic acidosis, haemolysis, hypotonia, seizures, liver failure | Neonatal iron storage disease Inborn error of bile acid synthesis | 24-48 hr |
| Alpha-1-antitrypsin deficiency Niemann-Pick type C | Neonate | ||
| Galactosaemia Tyrosinaemia Glycogen storage disease | Neonate – infant | ||
| Infant | Hypoglycaemia, jaundice, faltering growth, vomiting, hepatosplenomegaly, deranged LFT, chronic liver disease, fever, cataracts | Fructosaemia Cystic fibrosis PFIC 1, PFIC 2 Alpha-1-antitrypsin deficiency Alagille’s syndrome PFIC3 | InfantChild |
| Child | Hepatomegaly, faltering growth, developmental delay, short stature | Wilson’s disease* Autoimmune hepatitis* | Adolescent |
* Acute presentation with symptoms relating to acute liver failure may be due to an undiagnosed chronic condition, e.g. Wilsons disease or autoimmune hepatitis
LFT liver function test
PFIC progressive familial intrahepatic cholestasis
The Paediatric Acute Liver Failure (PALF) study group (http://www.palfstudy.org) reports that a cause for ALF is not determined in 47% of patients and that death or liver transplantation occurs in up to 45% [16]. At the author’s institution approximately 10%–20% of children with ALF will receive a liver transplant with a 1 year survival rate of 60%–75%.
Diagnosis of ALF
Fulminant hepatic failure often results in severe impairment of hepatic function or significant hepatocyte death in the absence of pre-existing liver disease with or without encephalopathy. In infants and young children the early stages of encephalopathy may be unrecognisable, in which case an escalating international normalised ratio (INR) along with an increasing activated partial thromboplastin time (APPT) and uncontrolled coagulopathy indicates a positive diagnosis. If a diagnosis of fulminant hepatic failure is strictly adhered to, a problem arises. It is difficult to distinguish between disease that is actually acute and that which is (undiagnosed) chronic disease, manifesting as acute. The two conditions can appear the same. A thorough investigation of all possible causes is necessary.
If onset is dramatic there is a possibility of the critical life threatening complication of cerebral oedema, requiring patients to be treated in the paediatric intensive care unit. If the child survives, liver function can return to near normal. However, increasing levels of bilirubin, INR and APPT, despite declining aspartate transaminase levels (which at this stage often indicates an accumulation of hepatocyte necrosis) suggest poor prognosis (Table 9.4). At the author’s institution some patients are offered alginate hepatocyte bead transplantation and are placed on the liver transplant waiting list. Ultimately patients with a poor prognosis are placed on the super urgent liver transplant list.
Table 9.4 Examples of biochemistry results pre and post orthotopic liver transplantation
| 2½ year old male with unknown aetiology | 10 year old female unknown aetiology | |||||||||
| 1 week | 1 week | |||||||||
| Day 1 | Day 2 | Day 3 | post OLT | Range | Day 1 | Day 3 | Day 5 | post OLT | Range | |
| Albumin | 31 | 25 | 24 | 46 | 35–55 g/L | 30 | 26 | 26 | 35 | 35-50 g/L |
| Bilirubin | 330 | 300 | 344 | 38 | <20 mmol/L | 268 | 291 | 327 | 47 | 3-20 mmol/L |
| AST * | 1140 | 328 | 474 | 80 | <77 IU/L | 1513 | 894 | 469 | 100 | 7-36 IU/L |
| GGT | 35 | 32 | 34 | 76 | <55 IU/L | 66 | 55 | 55 | 218 | 1-55 IU/L |
| ALP | 315 | 312 | 350 | 106 | <291 IU/L | 830 | 783 | 802 | 138 | 156-386 IU/L |
| ALT | 1763 | 944 | 750 | — | <55 IU/L | 1931 | — | 940 | — | 1-55 IU/L |
| INR | 3.04 | 7.6 | 8.75 | 1.10 | <1.2 (ratio) | 2.76 | 4.60 | 3.40 | 1.18 | < 1.2 (ratio) |
| APPT | 1.590 | 2.060 | 2.230 | 0.970 | <1.150 (ratio) | 1.51 | 1.60 | 1.65 | 1.02 | 0.85-1.15 (ratio) |
| Ammonia | 122 | — | 140 | — | <50 mmol/L | 108 | 126 | 164 | — | 12-50 mmol/L |
* A declining AST indicates hepatocellular necrosis
OLT orthotopic liver transplantation
AST aspartate transaminase
GGT gamma glutamyl transferase
ALP alkaline phosphatase
ALT alanine transaminase
INR international normalised ratio
APPT activated partial thromboplastin time
Table 9.4 illustrates the biochemical profiles of two different presentations of acute liver failure seen in a 2½-year-old boy and a 10-year-old girl. These examples show the blood biochemistry results in the days prior to orthotopic liver transplantation (OLT) and at 1 week post OLT, when there is a return to normal (or near normal) levels.
Nutritional requirements in ALF
If presentation is rapid, infants and children are often well nourished, so management is based on preventing hypoglycaemia and maintaining nutritional status until there is some clinical improvement (Table 9.5).
Table 9.5 Nutritional requirements in acute liver failure
| Energy |
|
| Glucose |
|
| Protein |
|
| Fat |
|
IMD inherited metabolic disorder
EAR estimated average requirement
MCT medium chain triglyceride
RNI reference nutrient intake
Dietetic management of ALF
The dietetic management of ALF lacks consensus [18]. Hypoglycaemia is present in 40% of patients with ALF due to increased plasma insulin levels secondary to reduced glucose uptake and gluconeogenesis [19]. Current practice is to provide maximum nutritional support during the diagnostic workup by avoiding hypoglycaemia and the build-up of toxic byproducts of metabolism. It is difficult to determine the diagnosis, particularly in infants, as IMD can be responsible and the liver may be sufficiently damaged to produce secondary biochemical abnormalities [20].
Older children and adolescents are less likely to have an undiagnosed IMD, with the exception of Wilson’s disease. A standard enteral formula may be given to these patients and if fluid restriction is no barrier nutritional requirements will be rapidly met.
Glucose management
In the presence of hypoglycaemia without a diagnosis, or if continuous feeding cannot achieve normoglycaemia, a continuous intravenous (IV) dextrose infusion is started. If there is no evidence of dehydration a two-thirds fluid restriction is imposed to avoid cerebral oedema. Sufficient energy must be given to avoid catabolism and its associated endogenous production of ammonia by delivering a glucose infusion rate according to age and weight (Table 9.6). The aim of treatment initially is to meet the glucose oxidation rate to prevent protein/fat catabolism.
Table 9.6 Emergency regimen
| Emergency regimen (ER) |
| On presentation an IMD may be suspected so an ER should be started as soon as possible as IV dextrose (and saline) with additional potassium if the child is having diuretic therapy. Enteral feeding should be started by adding glucose polymer (i.e. a lactose and fructose free source of carbohydrate) to either oral rehydration solution or water. Additional sodium/potassium supplements are necessary. The child should be fed continuously over 24 hours. |
| Guidelines for glucose requirements are: |
|
| Steps to take when initiating an ER |
| The amount of glucose given should be increased slowly according to tolerance Depending on the type of venous access and fluid allowance either 10%, 15% or 20% dextrose can be used In rare cases dextrose concentrations greater than 25% have been used to prevent hypoglycaemia |
| e.g. an infant, weight = 3.5 kg, fluid restriction = 67 mL/kg/day (i.e. ⅔ fluid restriction*) requiring 8 mg/kg/min glucose infusion will need: |
| 8.4 mL/hr (or 57.6 mL/kg) of 20% dextrose** via a central vein to meet glucose oxidation rate*** |
| Infants can tolerate up to 18 g glucose/kg/day, i.e. 12.5 mg/kg/min Initially, blood glucose monitoring should be hourly then increased to 4 hourly once stable Target serum blood glucose levels should be between 4-8 mmol/L |
* At the authors institution 100 mL/kg/day (or 4 mL/kg/minute) is a standard paediatric intensive care fluid allowance for children who are ventilated and sedated. A ⅔ fluid restriction equates to 67 mL/kg/day. If there is a high risk of cerebral oedema a maximum fluid allowance 67 mL/kg is imposed in children <1 year of age or ⅔ normal age appropriate fluid requirements for children >1 year.
** Dextrose concentrations greater than 10% will require central venous access to avoid the risk of thrombophlebitis.
*** At a 67 mL/kg fluid restriction, 57 mL/kg may not be available for dextrose as IV medications take precedence. IV medications are deducted from the total available IV fluid allowance before nutrition or maintenance fluids are added. A higher dextrose concentration at a lower volume may be required to meet the glucose infusion rate.
IMD inherited metabolic disorder
ER emergency regimen
IV intravenous
Usually a diagnosis of galactosaemia or tyrosinaemia is excluded quickly. Other conditions are likely to be ruled out from the clinical presentation. Following the use of the emergency regimen a suitable special formula can be used for the suspected condition, with little indication for the use of a modular feed. It is essential to meet basal protein and energy requirements as soon as possible to prevent catabolism to preserve muscle and fat stores. In the majority of cases a standard infant formula feed can be used and titrated against the IV dextrose over 24–48 hours to maintain blood glucose levels. In cases where fluid restriction is upheld, a glucose polymer may need to be added to the standard feed to provide sufficient glucose. In some institutions a modular feed is designed to provide complete nutrition (p. 184) .
Addition of protein
In the presence of encephalopathy it is important not to over-restrict protein and severe prolonged protein restriction is no longer practised as it could result in increased endogenous ammonia production from protein catabolism. However, a child with a suspected IMD of protein is initially protein restricted. With guidance from the metabolic team, complete protein requirements would be reintroduced within 24–48 hours. Further investigation by the metabolic team can provide an upper tolerable limit of the offending amino acid without increasing toxic byproducts. In practice a standard infant formula is used to provide up to the first 1 g/kg protein with the addition of a glucose polymer to meet the glucose oxidation rate. Infants should be given minimum protein requirements to meet the lower reference nutrient intake.
In cases where a modular feed is required protein should be added as soon as possible. If galactosaemia has not yet been excluded Essential Amino Acid Mix is a suitable lactose free source of protein. If tyrosinaemia has not been excluded, and depending on plasma amino acid levels, TYR Anamix can be given (the feed being based on a tyrosine free amino acid mix). The practice of using a methionine free formula has been replaced by the medication 2-(2-nitro-4-fluoromethylbenzoyl)-1,3-cyclohexanedione, which prevents the toxic build-up of succinyl acetone that can damage the liver.
Addition of fat
If a modular feed is used it will require the addition of fat to meet energy requirements. Fat in the form of long chain triglycerides (LCT), e.g. Calogen, or medium chain triglyceride (MCT) fat emulsions, e.g Liquigen, can be added as the condition of the child is monitored. Increments of 1 g fat/kg/day are recommended. If LCT fats are limited to <10% of total fat intake, small quantities of walnut oil (p. 601) may be needed to provide EFAs.
Vitamins and minerals
Many children will have adequate nutritional status during the acute phase and will probably receive adequate nutrition once they are established on a standard formula. In those who require a modular feed the addition of a complete vitamin and mineral supplement, such as Paediatric Seravit, is needed.
Feeding issues
In the majority of paediatric ALF cases medical treatment can result in full recovery. In children with advanced ALF nutrition support will be managed in the intensive care unit and adjustments to ventilation, sedation and fluid allowance will be needed. Often the rate at which any feed can be given is severely limited due to fluid restriction, with the majority of fluid volume taken up by IV medications and infusions. Continuous pump feeding together with concentrating the infant formula or adding glucose polymer is often required to avoid hypoglycaemia. In older children a 1.5–2 kcal (6–8 kJ)/mL formula may be required to meet energy demands. If hypoglycaemia is not corrected, partial or full IV dextrose infusion is required to meet glucose oxidation rates (Table 9.6). If renal filtration is commenced the fluid allowance may be liberalised to achieve an adequate feed rate.
In younger children after transplantation, surgery is performed to reroute the biliary ducts directly to the small bowel (i.e. a new Roux loop is formed) and a compulsory 5 day period of nil by mouth is usually imposed until the surgeons are satisfied that the risk of small bowel perforation is avoided. Parenteral nutrition (PN) is not usually given during this period unless nutritional status prior to OLT is very poor or, because of complications, the surgical team estimate that enteral feeding is unlikely to commence after 5 days. Older children can receive a duct-to-duct procedure, which does not involve rerouting of the biliary ducts to the small bowel, and feeding can usually commence within 24 hours.
The nutritional management of infants receiving liver transplantation due to ALF involves close nutritional monitoring for the first year. Some of these patients experience complications post liver transplant prolonging their progression to oral intake. In rare cases home enteral nutrition support may be required until appropriate weaning and adequate growth is achieved. Older children who have received a liver transplant for ALF tend to progress well on a normal diet once they get over the initial surgical insult. If normal growth is restored within the first year the need for specialist dietetic intervention diminishes.
A case study to show the management of a young child with ALF is given in Table 9.7.
Table 9.7 Case study – acute liver failure, unknown aetiology, suspected viral hepatitis
| Admission: 2 year 9 month old male twin, previously well and healthy Wt = 15.5 kg (83rd centile) MUAC = 17.2 cm (89th centile) Presentation history: Deranged liver function, coagulopathy and INR of 6.0 with acute liver failure, grade 1-2 encephalopathy, jaundice, pale stools, dark urine, lethargy, loss of appetite, abdomen soft, no organomegaly Impression: Post viral hepatic failure with encephalopathy and neutropenia Acute interventions: Admitted to PICU, intubated and sedated, ⅔ fluid restriction on 10% IV dextrose. Target glucose infusion rate = 7 mg/kg/min Clinical concerns: Blood glucose = 3.3 mmol/L. Bilirubin remained excessive, INR rapidly increased along with a declining AST indicating hepatocellular death. Albumin declined indicating worsening synthetic liver function. Required super urgent liver transplantation. |
Biochemistry (grey arrow depicting the time of liver transplant) 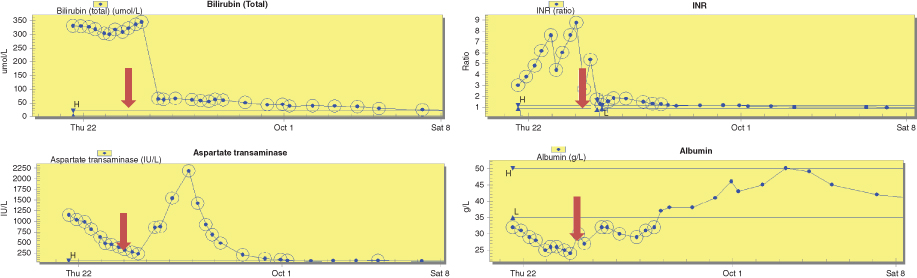 |
Nutrition management in PICU prior to transplant Day 1: Blood glucose levels (BM) were 3.3-4.1 mmol/L on ⅔ fluid restriction of 60 mL/kg. This comprised 43 mL/kg intravenous (IV) 10% dextrose and 17 mL/kg of IV medications in a 5% dextrose solution.
|
| Day 2: BM 4.7-5.4 mmol/L Aim to commence trophic nasogastric (NG) feeding, but due to limited volume to provide sufficient exogenous glucose to meet glucose needs and likely liver transplantation within 48 hours, enteral feeding was not initiated. Day 3: Received liver transplant during the early hours of the morning. Returned to PICU intubated and sedated. |
| Nutrition interventions post transplant – no special formula required Days 1-5: NBM due to new Roux loop. IV fluids met full fluid requirements. Extubated on day 2. Day 5: Weight = 14.7 kg (69th centile) MUAC = 16.3 cm (70th centile). Started NG feeding @10 mL/hr using a standard formula and increased feed rate 10 mL/hr every 6 hours. Target non ambulatory feed rate = 52 mL/hr which provided 80 kcal (335 kJ)/kg (EAR) + 2.2 g/kg protein. Pre surgical weight of 15.5 kg was used. Day 6: Transferred to high dependency unit on continuous NG feeding. Day 7: Chyle seen in abdominal drain fluid. Laboratory testing revealed chyle leak secondary to chylous ascites. Changed to high medium chain triglyceride (80% MCT) formula. Day 8: Started oral sips of fat free fluids and started low long chain triglyceride (LCT) diet. Day 10: Transferred to liver ward. NG feeding stopped during daytime hours to allow appetite for solid food. Continued overnight NG feeding. Days 12-14: Weight = 14.7 kg (67th centile) MUAC = 16.5 cm (75th centile). Chylous ascites resolved on day 14, NG tube removed. Started two oral nutritional supplement drinks (1.5 kcal, 6 kJ/mL) per day. |
| Week 3: Weight = 15.1 kg (74th centile) MUAC = 16.8 cm (81st centile). Energy supplements continued. Week 4: Weight = 15.4 kg (78th centile) MUAC = 17.2 cm (88th centile). Refused oral supplements but replaced these with high protein/energy snacks. Discharged to local hospital. Aim was to continue to provide up to 120% of EAR for energy over the next 3-6 months depending on growth and to monitor for excessive weight gain due to steroids. 3 month follow up at liver outpatient clinic: Weight = 16.0 kg (82nd centile) MUAC = 17.6 cm (92nd centile). Management unchanged. 6 month follow up at liver outpatient clinic: Weight = 16.7 kg (83rd centile) MUAC = 18.3 cm (96th centile). Had nonspecific unwell episodes which were related to sero-conversion of CMV. Symptoms resolved. 1 year follow up planned. 1 year post liver transplantation: Weight = 16.0 kg (53rd centile) MUAC = 17.6 cm (87th centile). Developed post transplant proliferative disorder (PTLD). Received bone marrow transplant at local hospital. Nutrition managed by dietitian at local hospital. Oral intake and weight reported to have improved. |
MUAC mid upper arm circumference INR international normalised ratio AST aspartate transaminase PICU paediatric intensive care unit NBM nil by mouth EAR estimated average requirement
Chronic liver disease
There are many causes CLD. Time of presentation can range from infancy through to adolescence; the chance of requiring liver transplantation increases with advancing age and liver disease progression. Children are at risk of malnutrition due to failing to meet the demands of growth. Post transplantation morbidity and mortality was strongly linked to advancing malnutrition status in a review at the author’s institution. Common factors contributing to malnutrition in ESLD are listed in Table 9.8.
Table 9.8 Potential causes of malnutrition in chronic liver disease
| Physical complications | Inadequate intake, anorexia, nausea, vomiting, irritability |
|
| Malabsorption | Impaired nutrient digestion and absorption |
|
| Hypermetabolic effects | Increased nutritional requirements |
|
| Medication effects | Volume displacement, lethargy, timing of medication interfering with meal times |
|
Nutritional requirements
Nutritional requirements in CLD are given in Table 9.9. The dietary management of disorders that lead to CLD are given below.
Table 9.9 Nutritional requirements for chronic liver disease
| Acute on chronic liver disease with cholestasis | |||
| Energy | infants 100-150 kcal (420-630 kJ)/kg/d depending on level of malabsorption | ||
| children 100-120% EAR | |||
| Protein | 10-12% TE | ||
| Fat | 1-4 g/kg of which 30-70% as MCT (dependent upon diagnosis) | ||
| Chronic liver disease | |||
| Energy | Protein | Fat | |
| Infants (0-1 year) | 100-150 kcal (420-630 kJ)/kg/day | 1-4 g/kg (10-12% TE) | meet EAR – if |
| Children (pre school) | 130-150% EAR | 3-6 g/kg | cholestasis use |
| Children (school age) | 100-130% EAR | 3-5 g/kg | high MCT formula |
| ESLD all ages | 150% EAR | up to 4 g/kg | or supplement |
EAR estimated average requirement [17] MCT medium chain triglycerides TE total energy ESLD end stage liver disease
Disorders presenting in infancy
- inherited metabolic disorders, e.g. glycogen storage disease, urea cycle defect
- infections, e.g. hepatitis
- biliary malformations, e.g. biliary atresia, choledocal cysts
- vascular lesions, e.g. hepatocellular carcinoma, haemangioma
- toxic and nutritional disorders
- cryptogenic disorders, e.g. cirrhosis or hepatitis
Jaundice
Jaundice is a condition that causes the infant or child to appear yellow in complexion and in the whites of their eyes; it is classified as either conjugated or unconjugated. Unconjugated jaundice is characterised clinically by a jaundiced appearance without bilirubin in the urine. This may be physiological in the newborn due to an increase of bilirubin production, decreased bilirubin clearance or a combination of both. Treatment does not usually require dietetic intervention. Conjugated jaundice occurs when the total serum bilirubin is raised and the conjugated fraction is >20% of the total bilirubin (normally <5%). This needs further investigation and usually requires dietetic management. Conjugated bilirubin is made water soluble by the addition of glucuronide in the liver and enters the bile. If bile flow from the liver is reduced the stools will lack pigment. In this case the (conjugated) bilirubin glucuronide passes into the serum and is then excreted as dark urine.
Infantile conjugated hyperbilirubinaemia
Infantile CHB is the most common presentation for an infant with hepatobiliary disease. The majority of newborn infants seen in specialist liver centres present with CHB. This type of jaundice represents significant hepatobiliary disease and is described as cholestatic liver disease. The bile flow from the liver into the gut is limited and fat emulsification and digestion is reduced. This leads to malabsorption of fat, fat soluble vitamins and some minerals. Steatorrhoea, growth failure and rickets are common clinical consequences [21, 22]. Galactosaemia needs to be excluded as a diagnosis. Dietetic management of CHB is summarised in Table 9.10. Most of the possible diagnoses for CHB are given below.
Table 9.10 Summary of dietetic management for infantile conjugated hyperbilirubinaemia/cholestatic jaundice
1. Is galactosaemia suspected?
|
2. Is galactosaemia diagnosed?
|
3. Is infant breast fed?
|
4. Is cholestasis is resolving?
|
5. Are blood sugar levels maintained?
|
6. Is there faltering growth with an adequate feed volume?
|
7. At weaning
|
| 7. Continue to review and monitor |
MCT medium chain triglyceride EFA essential fatty acids
Common liver diseases in infants and young children and dietetic management
Neonatal hepatitis
The cause of neonatal hepatitis is unknown. Severity varies and rarely results in cirrhosis. Usually bile flow resolves with time, but in a majority of these cases fat malabsorption is treated with formulas containing a high percentage of MCT until bile flow and adequate growth resume. Suitable formulas with a high MCT content are given in Table 9.11.
Table 9.11 Formulas and supplements high in medium chain triglycerides per 100 mL
| % MCT | Energy kcal (kJ) | CHOg | Proteing | Fatg | Linoleicacidmg | α-linolenic acid mg | EFA ratio | OsmolalitymOsm/kgH20 | Dilution% | |
| Powdered formulas | ||||||||||
| Caprilon (SHS)* | 75 | 66 (275) | 7.0 | 1.5 | 3.6 | 5.6 | 0.8 | 7.5 | 233 | 12.7 |
| Heparon Junior (SHS) | 49 | 86 (360) | 11.6 | 2.9 | 3.6 | 8.2 | 1.2 | 6.8 | 310 | 18.0 |
| Generaid Plus (SHS) | 32 | 102 (425) | 13.6 | 2.4 | 4.2 | 3.7 | 1.0 | 4 | 390 | 22.0 |
| Pregestimil Lipil (Mead Johnson)* | 55 | 68 (285) | 6.9 | 1.89 | 3.8 | 10.1 | 0.6 | 16.8 | 330 | 13.5 |
| Pepti-Junior (Cow & Gate)* | 50 | 66 (275) | 6.8 | 1.8 | 3.5 | 12.8 | 0.2 | 64 | 210 | 12.8 |
| MCT Pepdite (SHS)* | 75 | 68 (285) | 8.8 | 2.0 | 2.7 | 5.1 | 0.8 | 6.4 | 290 | 15.0 |
| Monogen (SHS)* | 80 | 74 (310) | 12 | 2.0 | 2.1 | 1 | 0.2 | 4.6 | 280 | 17.0 |
| Peptamen Junior (Nestle) | 60 | 100 (420) | 13.8 | 3.0 | 3.85 | — | — | — | 260 | 22.0 |
| Emsogen (SHS)** | 83 | 88 (370) | 12 | 2.5 | 3.3 | 3.4 | 0.1 | 48 | 580 | 20.0 |
| Ready to feed formulas | ||||||||||
| Peptamen Junior Liquid | 60 | 100 (420) | 13.2 | 3.0 | 4.0 | — | — | — | 319 | — |
| (Nestle) | ||||||||||
| Peptamen Junior Advance | 61 | 150 (630) | 18 | 4.5 | 6.6 | — | — | — | 380 | — |
| (Nestle) | ||||||||||
| Nutrison MCT (Nutricia)*** | 61 | 100 (420) | 12.6 | 5.0 | 3.3 | 5.3 | 0.7 | 7.6 | 265 | — |
| Nutrison Peptisorb | 50 | 100 (420) | 17.6 | 4.0 | 1.7 | — | — | — | 455 | — |
| (Nutricia)*** | ||||||||||
| Paediasure Peptide (Abbott) | 50 | 100 (420) | 13.0 | 3.0 | 4.0 | — | — | — | 272 | — |
| Energy modules | ||||||||||
| Liquigen (SHS) | 98 | 450 (1880) | 0 | 0 | 50 | 0 | 0 | 0 | 10 | — |
| MCT oil (SHS) | 99 | 855 (3575) | 0 | 0 | 95 | 0 | 0 | 0 | — | — |
| Calogen (SHS) | 0 | 450 (1880) | 0.1 | 0 | 50 | 24 | 4.7 | 5 | 360 | — |
| Duocal (SHS) | 35 | 123 (515) | 18.1 | 0 | 5.6 | 4.4 | 1.1 | 4 | 196 | 25.0**** |
| Liquid Duocal (SHS) | 28 | 166 (695) | 23.7 | 0 | 7.9 | 15.7 | 0.2 | 68 | 400 | — |
| MCT Duocal (SHS) | 75 | 165 (690) | 24 | 0 | 7.7 | 6.3 | 0.9 | 7 | 427 | 33.0**** |
| Liquid Maxijul | 0 | 247 (1030) | 61.9 | 0 | 0 | 0 | 0 | 0 | 1400 |
* Infant formulas
** Formulas not suitable for children <5years old
*** Formulas not suitable for children <7 years old
**** Suggested dilution
MCT medium chain triglyceride
EFA essential fatty acids
SHS Scientific Hospital Supplies

Full access? Get Clinical Tree


