Fig. 10.1
Overexpression of HSP70 affects multiple cancer-relevant pathways. (a) HSP70 inhibits the extrinsic apoptosis pathway by binding to the death receptors DR4 and DR5, and preventing the formation of the DISC. HSP70 inhibits the intrinsic pathway by inhibiting ASK1 and JNK, thereby preventing BAX and BID translocation to the mitochondria. HSP70 also prevents apoptosis downstream of the mitochondria by binding to APAF-1 and preventing formation of the apoptosome. In addition, HSP70 stabilizes lysosome membranes and inhibits lysosome membrane permeabilization (LMP). (b) HSP70 inhibits both p53-dependent and -independent senescence. (c) As an obligate co-chaperone for HSP90, HSP70 is essential for the proper functioning of HSP90, and proper folding of HSP90 client proteins
10.3.2 Senescence, Lysosome Function
In addition to suppressing key proteins in the apoptotic pathway, evidence suggests that HSP70 can also inhibit senescence. For example, siRNA-mediated depletion of HSP70 results in a senescent phenotype, including flattened cell morphology, increased phosphorylation on serine 15 of p53, decreased proliferation, and increased senescence-associated β-galactosidase staining (Yaglom et al. 2007). Additionally, expression of oncogenic forms of PI3K and RAS in breast epithelial cells showed dependency on HSP70 for transformation, in both a p53-dependent and -independent manner, respectively (Fig. 10.1b) (Gabai et al. 2009). Lysosome stability is important for tumor growth, and lysosomal membrane permeabilization (LMP) induced by various stresses results in cell death, while extracellular release of lysosomal proteases can promote tumor invasion (Bivik et al. 2007; Doulias et al. 2007; Dudeja et al. 2009; Zorzi and Bonvini 2011). Specifically in cancer but not normal cells, HSP70 has been found bound to the lysosomal membrane, stabilizing it in the face of stressors such as tumor necrosis factor (TNF), etoposide, UV radiation, and hydrogen peroxide; notably, when HSP70 is depleted, these stresses result in increased cell death (Nylandsted et al. 2004; Bivik et al. 2007; Doulias et al. 2007). In addition, HSP70 interacts with proteins involved in lysosomal membrane permeabilization (LMP), including BAX, JNK, and p53, and this protein can prevent LMP and the release of proteolytic hydrolases, and to thus maintain cellular integrity.
10.3.3 HSP90
The chaperones HSP70 and HSC70 are obligate co-chaperones for HSP90. Not surprisingly then, depletion or inhibition of HSP70 results in reduced solubility of HSP90 client proteins, which become sequestered and inactivated in detergent insoluble compartments in the cell (Leu et al. 2011; Murphy 2013). In a well-controlled study by Workman and colleagues, simultaneous silencing of HSP70 and heat shock cognate 70 (HSC70) resulted in proteasome-dependent degradation of the HSP90 cancer-critical client proteins, C-RAF and CDK4, and initiation of apoptosis in cancer cells, but not in normal non-transformed cells (Powers et al. 2008). These data argue that simultaneous targeting of both HSP70, and the constitutively expressed family member HSC70, may be required for effective cancer therapy.
10.4 HSP70 Chaperone Structure
As indicated above, HSP70 is implicated in the pathogenesis of cancer (Ciocca and Calderwood 2005; Powers et al. 2009) and neurodegenerative diseases (Patury et al. 2009; Evans et al. 2010). Therefore, targeting HSP70 function represents a novel therapeutic strategy for these diseases. This has led to increased interest in understanding the structure and mechanism of allostery of this chaperone. The Hsp70 family of proteins (HSP70 in humans, DnaK in E. coli) has a highly conserved domain structure. Each contains a 45 kDa N-terminal nucleotide binding domain (NBD) that has ATPase activity and a 25 kDa C-terminal protein substrate binding domain (SBD), joined by a short flexible linker (Bukau and Horwich 1998; Mayer 2010; Zuiderweg et al. 2013) (Fig. 10.2a). The interdomain linker is only 12 residues in length and is highly conserved (Vogel et al. 2006). Because these two domains are rarely seen together in crystal structures, their detailed structures were described separately in the 1990s. The NBD consists of two flexible lobes, I and II, and each is further divided into subdomains A and B, which form a deep nucleotide-binding cleft (Bork et al. 1992). Structural data suggest that the nucleotide-binding site is located at the bottom of the cleft (Flaherty et al. 1990). This structure was first reported for the NBD isolated from bovine ADP-bound HSC70, and subsequently has been confirmed by others (Flaherty et al. 1990; Harrison et al. 1997; Sriram et al. 1997; Chang et al. 2008).
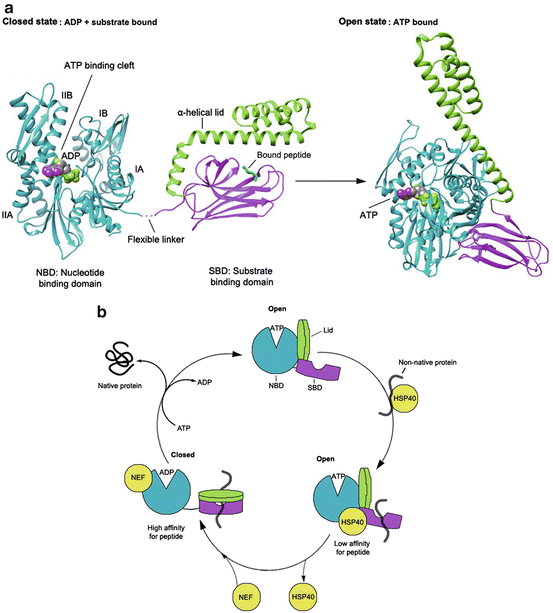
Fig. 10.2
Structure and allosteric cycle of HSP70. (a) The proposed confirmations of the two ‘end-point’ states of HSP70: the closed state is illustrated by the crystal structure of ADP-bound DnaK (Escherichia coli HSP70; PDB 2KHO; right panel); the open state is illustrated by the crystal structure of the ATP-bound state of Sse1 (yeast Hsp110; PDB 2QXL; left panel). (b) ATP binding to the nucleotide binding domain (NBD) facilitates low affinity binding of substrates, such as those presented to HSP70 via the HSP40 co-chaperone. HSP40 stimulates nucleotide hydrolysis and increases the affinity for substrate, by mediating the closure of the SBD α–helical lid. Nucleotide Exchange Factors (NEFs) assist with ADP release, which causes substrate release
The substrate binding domain (SBD) consists of two functionally relevant subdomains: SBDβ, a 15 kDa β-sandwich subdomain comprised of two four-stranded β sheets with a hydrophobic pocket for peptide binding between them, and SBDα, a 10 kDa mobile α-helical C-terminal region that is believed to function as a “lid” that closes over the substrate (Wang et al. 1998) (Fig. 10.2a). This structure was first described in E. coli for the SBD of DnaK in complex with a short peptide (Zhu et al. 1996). C-terminal to the “lid” is the least conserved region amongst HSP70 family members, which terminates with an EEVD (amino acid designations) motif, capable of interacting with tetratricopeptide repeat (TPR) domain-containing proteins. Sequence variation of this region is believed to allow different HSP70 family members to interact with distinct co-chaperones.
10.5 The HSP70 Functional Cycle
HSP70 assists the folding of its clients using repeated cycles of binding and releasing of misfolded proteins. The ability of the SBD to preferentially bind to exposed hydrophobic polypeptide sequences allows this domain to recognize the non-native states of many proteins (Chiti and Dobson 2006; Eichner and Radford 2011). In some cases, members of the DnaJ/HSP40 family of co-chaperones recognize and bind to such substrates first, and then present them to the HSP70 protein (Bukau and Horwich 1998; Han and Christen 2003). Similar to other ATP-dependent chaperones, the activity of HSP70 requires the energy of ATP binding and hydrolysis and involves critical allostery between the NBD and SBD. Specifically, ADP-ATP exchange in the NBD, which is catalyzed by nucleotide exchange factors (NEFs) like BAG-1 (Liu and Hendrickson 2007; Schuermann et al. 2008), triggers the attachment of the interdomain linker and the α-helical “lid” to the NBD, thereby opening up the peptide binding pocket (this is referred to as an “open” domain-docked conformation of HSP70) (Fig. 10.2b). Subsequently, this results in release of bound substrate from the SBD. Released unfolded peptides are believed to then spontaneously collapse into their native state in free solution (Sharma et al. 2010), or to re-bind to HSP70 for further unfolding, or to transfer to downstream chaperones and possibly be targeted for degradation. Binding of a new peptide to the SBD induces a conformational change that is propagated back to the NBD, increasing the rate of ATP hydrolysis. Finally, hydrolysis of ATP is signaled back to the SBD, resulting in closing of the α-helical “lid” and enhancement of substrate affinity (that is, the “closed” domain-undocked conformation of HSP70) (Fig. 10.2b).
In the attempt to understand the allosteric regulation of HSP70, researchers have focused on comparing the crystal structures of ADP- and ATP-bound DnaK, an HSP70 orthologue from E. coli that shares about 60 % sequence homology with human HSP70 and has a similar domain structure. These studies revealed that in ADP-bound DnaK, the NBD and SBD behave as independent units, and the structures observed show no stable communication between them (Swain et al. 2007; Bertelsen et al. 2009) (Fig. 10.2a, left panel). However, upon ATP binding, the protein undergoes large conformational changes in both domains, which come together to form an intimately packed domain-docked structure (Fig. 10.2a, right panel) (Kityk et al. 2012; Zhuravleva et al. 2012; Qi et al. 2013). It was shown that the interdomain linker of DnaK binds between the IA and IIA subdomains of the isolated NBD, and this alone is necessary and sufficient for ATPase activation (Vogel et al. 2006; Swain et al. 2007). Similarly, the recently described structure of an HSP70 homolog, yeast HSP110, Sse1, shows that the linker is hidden between the NBD and SBD when HSP70 is in the ATP-bound state. This suggests a tight NBD-SBD interaction and is consistent with previously observed ATP-mediated conformational changes (Buchberger et al. 1995; Rist et al. 2006; Liu and Hendrickson 2007; Mapa et al. 2010). These structural data highlight the role of the interdomain linker in the docking of the SBD onto the NBD; however they do not explain how binding of ATP induces allosteric “lid” opening and subsequent loss of affinity for substrates (Flynn et al. 1989; Schmid et al. 1994). To understand the mechanism of this allosteric signal transmission between the NBD and SBD, Mayer and colleagues captured and characterized a normally transient conformation of human HSP70 bound to ATP by engineering disulfide bonds between SBDα and the subdomain IB of the NBD. They showed that ATP binding to the N-terminus of HSP70 induces a clockwise rotation of NBD lobe II, leading to closure of the nucleotide-binding cleft between subdomains IB and IIB. This conformational change widens a space between subdomains IA and IIA, creating a binding site for the interdomain linker, which then docks SBDβ to the ATP-stabilized NBD. The intimate interaction between the NBD and SBDβ interfaces results in SBDα displacement from SBDβ and binding to subdomain IB of the NBD (“lid” opening). Conformational changes of the SBD also propagate to the other side of SBDβ (loop L3,4 of the β-domain), where the substrate-binding cleft opens up, allowing bound substrates to rapidly dissociate. These dramatic allosteric rearrangements in both domains triggered by ATP binding lead to conversion of HSP70 into the stable, open domain-docked state (Kityk et al. 2012; Zhuravleva et al. 2012; Qi et al. 2013). This study reveals that open (ATP-bound) conformation of HSP70 differs from the closed (ADP-bound) not only in the orientation of the α-helical lid, but also in the conformation of the peptide-binding pocket of SBDβ. It also identifies the NBD-linker interaction as a first critical step in interdomain communication. Combined with the knowledge that mutations in the linker abolish chaperone function of HSP70 (Han and Christen 2001), these data point to a role for the linker as a key site in allosteric regulation.
10.6 Opportunities to Interfere with the HSP70 Function
Increased understanding of how ligands modulate the HSP70 allosteric cycle facilitates the design of small allosteric inhibitors of HSP70 for therapeutic targeting of this chaperone. ATP hydrolysis plays a significant role in regulating the HSP70 chaperone activity. Therefore, inhibition of the enzymatic activity of HSP70 with competitive nucleotide analogs that disrupt HSP70-ATP interaction represent a logical approach to interfere with the function of this chaperone. While this strategy has been successful for HSP90 (Messaoudi et al. 2008), compounds that bind directly to the ATPase domain of HSP70 were not identified until recently. A lack of progress in targeting this domain can be explained by its unique structural and chemical characteristics. HSP70 has a 300-fold higher affinity for nucleotide binding when compared to HSP90 (Borges and Ramos 2006; Williamson et al. 2009; Massey 2010). In addition, analysis of the ATP-site, combining structure-based design and computational modeling, revealed that the ATP-binding site of HSP70 consists predominantly of hydrophilic residues, and that the interaction energy between the nucleotide and the chaperone is mostly mediated by the phosphate groups (Hurley 1996; Liu and Hendrickson 2007). This is generally associated with poor druggability of the enzyme active site because it does not allow the formation of hydrogen bonds and hydrophobic interactions between most inhibitors and the ATP-binding pocket (Halgren 2009). Despite these barriers, the ATP-binding site of HSP70 has a favorable size and is well enclosed, indicating that it remains a potentially druggable site. Indeed, recently novel competitive inhibitors of the HSP70 ATPase activity were identified, and these inhibitors were shown to inhibit cancer cell viability in vitro (Williamson et al. 2009).
Another way to modify HSP70 activity is by inhibiting its interactions with important co-chaperones, such as J proteins, nucleotide exchange factors (NEFs) and TPR domain-containing proteins. This can be achieved by blocking protein-protein interactions (PPIs) between HSP70 and co-chaperones or by targeting allosteric sites that disrupt these interactions. Normally, binding of a particular co-chaperone to HSP70 results in activation of a specific function in chaperone biology such as protein trafficking and translocation, protein folding or degradation (Table 10.1). Consequently, the rationale for developing chemical modulators of these interactions is to target a specific subset of HSP70 biology without impacting global proteostasis. Although PPIs are very difficult to inhibit (Arkin and Wells 2004; Gestwicki and Marinec 2007), several molecules have already been reported to specifically disrupt the interaction of HSP70 with particular co-chaperones and lead to distinct effects on cellular homeostasis (Yi and Regan 2008; Roodveldt et al. 2009; Dorard et al. 2011). Taken together, the available structural data suggest there are many opportunities to chemically target HSP70. Recent promising advances in developing HSP70 inhibitors are discussed below.
Table 10.1
Essential components of the HSP70 chaperone machinery
Co-chaperones | Function |
|---|---|
J domain proteins | 1. These interact with HSP70 at lobes IA and IIA of the Nucleotide Binding Domain (NBD) and the adjacent linker region to stimulate ATP hydrolysis and subsequently increase affinity for substrate binding by HSP70 |
2. J proteins can specifically recognize and bind misfolded substrates and present them to HSP70 for refolding | |
These interact with HSP70 at lobes IB and IIB of the NBD to catalyze the exchange of ADP to ATP, which results in lid opening and substrate release | |
Nucleotide Exchange Factors (NEFs) | HSP110 has a similar domain structure to HSP70. It functions as a “holdase” for non-native proteins |
Hsp110 | Interacts with lobe II of the NBD of HSP70 and releases nucleotide upon binding |
HSPBP1 (Hsp70 binding protein 1) | Contains a C-terminal “BAG” domain that binds to lobes IB and IIB of the NBD of HSP70, which causes ADP release |
BAG (BCL-2-associated AthanoGene) domain proteins | Has a ubiquitin-like domain that regulates the fate of HSP70-bound substrates |
BAG-1, Bcl-2-associated athanogene 1; | Substrate-specific regulation of HSP70 clients for inhibition or increase of proteasomal degradation |
BAG-2/3, Bcl-2-associated athanogene 2/3 | Induced by HSF1, may play a role in tumor formation, stabilizes oncogenes and suppresses apoptosis in complex with HSP70 |
Tetratricopeptide repeat (TPR) domain-containing proteins | Bind to the C-terminus of HSP70 at the EEVD motif |
CHIP | A ubiquitin E3 ligase, directs ubiquitination of HSP70-bound substrates for proteasome degradation |
HOP | Mediates the association of HSP70 and HSP90, thereby allowing transfer of substrates |
10.7 HSP70 Inhibitors
Recently, several inhibitors of HSP70 have been reported. Some of the most promising ones are discussed below, and their structures are depicted in Fig. 10.3a. For a more detailed comparative analysis of all the HSP70 inhibitors, readers are directed to some excellent recent reviews on this subject (Patury et al. 2009; Evans et al. 2010; Powers et al. 2010; Goloudina et al. 2012; Murphy 2013).
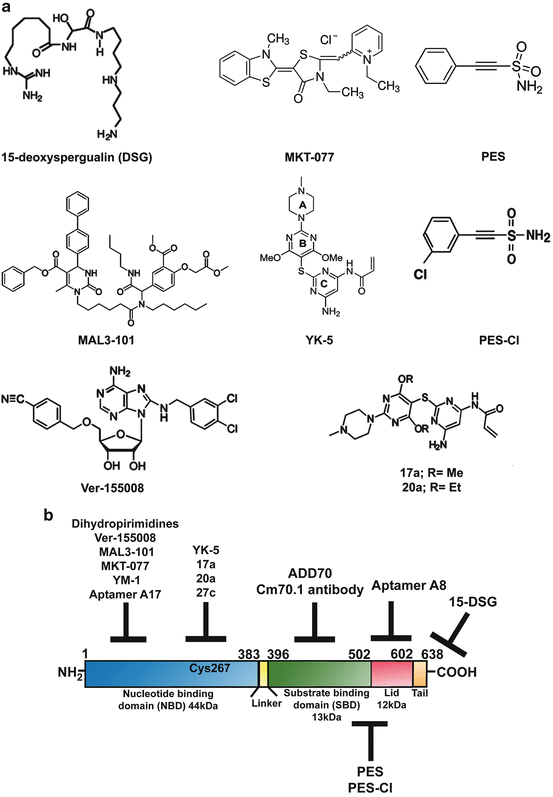
Fig. 10.3
Small molecule inhibitors of HSP70. (a) Chemical structures of HSP70 inhibitors. (b) Schematic representation of HSP70 domain architecture and the possible sites targeted by the various described inhibitors
10.7.1 Deoxyspergualin/Dihydropyrimidines
One of the first compounds identified to bind and inhibit HSP70 function was the antibiotic 15-deoxyspergualin (DSG) (Plowman et al. 1987). DSG binds to the C-terminus of HSP70 (Fig. 10.3b) with a K D of 4 μM and hinders its function by interfering with its steady-state ATPase activity (Nadler et al. 1995; Brodsky 1999). Although this compound showed promise as an antitumor agent in a mouse leukemia model (Plowman et al. 1987), its efficacy as an anticancer drug in a clinical trial involving metastatic breast cancer was shown to be limited (Dhingra et al. 1994). A subsequent search for compounds with a structural similarity to DSG led to the identification of a class of dihydropyrimidines that block HSP70 ATPase activity (Fewell et al. 2001, 2004). This second-generation of dihydropyrimidines, called the MAL3 series of compounds, were also developed through a screening process (Fewell et al. 2004). Among them, MAL3-101 deserves special mention. It interferes with the co-chaperone HSP40’s ability to stimulate the ATPase activity of HSP70 and prevents its function (Rodina et al. 2007). MAL3-101 blocks cancer cell proliferation (Braunstein et al. 2011) and was shown to be efficacious in a xenograft model of multiple myeloma (Rodina et al. 2007; Braunstein et al. 2011). This series of compounds awaits further testing as prospective anti-cancer drugs.
10.7.2 MKT-077
MKT-077, a rhodocyanine compound, was discovered to accumulate selectively in mitochondria of tumor cells and shown to exert an anti-proliferative effect on cancer cells (Koya et al. 1996). Interestingly, this compound binds an allosteric negatively charged pocket close to the ATP-binding site on HSP70 (Fig. 10.3b), and inhibits the ADP-bound state of this protein, interfering with the HSP70 folding cycle (Rousaki et al. 2011). MKT-077 is primarily toxic to tumor cells and its mechanism of action involves blocking the mitochondrial form of HSP70, HSP70-9 (also called Grp75) (Wadhwa et al. 2000; Rousaki et al. 2011). Mouse xenograft studies demonstrated time- and concentration-dependent antitumor activity for this compound (Chiba et al. 1998). In addition, MKT-077 is cytotoxic to different tumor cell lines, inhibits autophagy and reduces the level of HSP90 client proteins, such as CDK4 and HER-2 (Budina-Kolomets et al. 2014). However, analysis of this compound in a Phase I clinical trial led to severe renal impairment and toxicity in both animal and human subjects, thus limiting further testing and use of this compound (Propper et al. 1999). Recently, a derivative of MKT-077, called YM-1 was developed and shown to be selectively toxic to a variety of tumor cell lines, and to restore tamoxifen sensitivity to tamoxifen-resistant MCF-7 cells (Koren et al. 2012). Thus YM-1 has promising therapeutic potential.
10.7.3 Allosteric Inhibitors of HSP70: Compounds YK-5, 17a, 20a and 27c
Chiosis and colleagues recently identified a previously unrecognized allosteric site in HSP70 using a computational approach (Rodina et al. 2013). Analysis of the geometry and the environment of this computationally-identified allosteric site provided an initiation point for the development of ligands that would bind to this site. The first allosteric inhibitor designed and tested in this series was YK-5, which was found to block both HSP70 and HSC70 function (Rodina et al. 2011). YK-5 blocks HSP70 function by altering the interactions between HSP70, HSP90 and HOP, thus inhibiting the formation of this chaperone complex. This leads to the destabilization of the HSP90 client proteins HER2, RAF-1 and AKT, thereby leading to their degradation. YK-5 reportedly blocks cell proliferation, induces apoptosis and prevents HSF-1 activation or feedback heat shock response in cancer cells. However, YK-5 was found to be an irreversible covalent modifier of HSP70; therefore, this group further developed this series of inhibitors, leading to the identification of two irreversible derivatives, 17a and 20a, which selectively bind HSP70 in cancer cells. Addition of 17a and 20a at low micromolar doses led to a reduction in the steady state levels of HSP70-HOP-HSP90 chaperone complex and the degradation of HSP90 client proteins, along with the induction of cell cycle arrest and apoptosis (Rodina et al. 2013; Kang et al. 2014).
10.7.4 Peptide Inhibitors
HSP70 has a prominent cytoprotective role and this protein increases the oncogenic potential of cancer cells and blocks apoptosis by interacting directly with, and neutralizing the function of, apoptotic protease activation factor-1 (APAF-1) (Beere et al. 2000) and apoptosis-inducing factor (AIF) (Ravagnan et al. 2001). Garrido and co-workers utilized this anti-apoptotic characteristic of HSP70 and designed the peptide ADD70 (AIF-Derived Decoy for HSP70), which is both nontoxic and cytosolic, and is able inhibit the interaction of HSP70 with AIF and other client proteins. They employed deletion mutants and computer modeling methods to develop this AIF-derived blocking peptide, corresponding to the amino acids 150–228 of AIF, a region necessary for binding the SBD of HSP70 (Fig. 10.3b) and neutralizing its activity (Schmitt et al. 2003). Further investigation by this group showed that expression of ADD70 peptide in tumor cells decreased their tumorigenicity by increasing infiltration of cytotoxic CD8+ T cells into the tumor. In addition, ADD70 sensitized rat colon cancer cells and mouse melanoma cells to the chemotherapeutic agent cisplatin (Schmitt et al. 2006), indicating that it shows promise as an anti-tumor agent. Due to the large size of the ADD70 peptide, introducing it into tumor cells can be sometimes challenging. To eliminate this limiting factor this group developed smaller peptide aptamers, A8 and A17, which bind to the peptide-binding and the ATP-binding domains of HSP70, respectively, and specifically inhibit the chaperone activity of HSP70. A 13-amino acid peptide was further synthesized from the variable region of A17 (called P17) that specifically blocked HSP70 and induced regression of subcutaneous tumors in vivo, via the recruitment of macrophages and T lymphocytes into the tumor bed (Rerole et al. 2011).
10.7.5 Ver-155008
Massey and co-workers developed the first adenosine-derived inhibitor of HSP70 by designing ATP-analogs that bind the ATP-binding pocket of the chaperone by using the crystal structure of HSC70/BAG-1. This led to the development of the ATP-analog Ver-155008, which binds to HSP70 with a K D of 0.3 μM and inhibits its activity (Williamson et al. 2009). Ver-155008 is exclusively cytotoxic to cancer cells and downregulates expression of the HSP90 client proteins CDK4, HER2 and RAF-1 in tumor cells (Massey et al. 2010; Budina-Kolomets et al. 2014). It also has anti-proliferative function in a variety of cancer cells; however its ability to induce apoptotic cell death in cancer cells is somewhat limited (Massey et al. 2010). It remains to be seen if derivatives of Ver-155008 can be developed to increase the efficacy of this compound.
10.7.6 The HSP70 Antibody Cm70.1
HSP70 was first reported by Multhoff and co-workers to be localized in the plasma membrane of tumor cells (Multhoff et al. 1995; Multhoff and Hightower 1996), where it is integrated into lipid rafts (Schilling et al. 2009). Although HSP70 lacks a transmembrane domain, its presence in the plasma membrane is believed to be necessary for the stabilization of tumor cell membranes from damage due to environmental stress. Intriguingly, only tumor cells, but not corresponding normal cells, express HSP70 on the surface and stain positively for the IgG1 mouse monoclonal antibody (mAb) cmHsp70.1 (Multhoff and Hightower 2011). The Cm70.1 antibody specifically recognizes an epitope in the C-terminus of the inducible HSP70 (Fig. 10.3b) in viable tumors of both humans in vitro and in mouse tumors in vivo (Stangl et al. 2011). Extensive screening studies in nearly 1,000 primary human tumor biopsies and the corresponding normal tissues indicate that HSP70 is often expressed on the plasma membrane of tumor cells only, and its presence is linked with decreased overall survival of patients; therefore Cm70.1 serves as a negative prognostic marker (Pfister et al. 2007). Interestingly, injecting tumor bearing mice with the cmHsp70.1 antibody significantly inhibited tumor growth and enhanced overall survival, while injection of the peptide corresponding to C-terminal region (aa 450–461) of HSP70 abrogated this effect. This finding suggests that cmHsp70.1 may be a useful therapeutic tool.
Related posts:
 Introduction to Cell Stress Responses in Cancer: The Big Picture
Introduction to Cell Stress Responses in Cancer: The Big Picture
 Pro-senescence Therapy for Cancer: Time for the Clinic
Pro-senescence Therapy for Cancer: Time for the Clinic
 Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
Targeting Hypoxic Adaptations of Cancer Cells: Molecular Mechanisms and Therapeutic Opportunities
 p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
p53 at the Crossroads Between Stress Response Signaling and Tumorigenesis: From Molecular Mechanisms to Therapeutic Opportunities
 Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
Cell Cycle Checkpoint and DNA Damage Response Defects as Anticancer Targets: From Molecular Mechanisms to Therapeutic Opportunities
 Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Cell-Nonautonomous ER Stress-Mediated Dysregulation of Immunity by Cancer Cells
Stay updated, free articles. Join our Telegram channel

Full access? Get Clinical Tree


